


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Mechanism of action: Dostarlimab is a humanised mAb of the IgG4 isotype that binds to PD-1 receptors and blocks the interactions of binding with its ligands PD-L1 and PD-L2. The inhibition of PD-1 pathway-mediated immune response results in inhibition of T-cell function such as proliferation, cytokine production, and cytotoxic activity. Dostarlimab potentiates T-cell responses, including anti-tumour immuno responses through blockade of PD-1 binding to PD-L1 and PD-L2. In syngeneic mouse tumour models, blocking PD-1 activity resulted in decreased tumour growth.
Clinical efficacy and safety: The efficacy and safety of JEMPERLI were investigated in the GARNET study, a multicentre, uncontrolled, multiple parallel cohort, open-label study. The GARNET study included expansion cohorts in subjects with recurrent or advanced solid tumours who have limited available treatment options. Cohort A1 enrolled patients with mismatch repair deficient (dMMR)/microsatellite instability-high (MSI-H) EC who have progressed on or after a platinum-containing regimen.
Patients received 500 mg dostarlimab every 3 weeks for 4 cycles followed by 1000 mg dostarlimab every 6 weeks. Treatment continued until unacceptable toxicity or disease progression for up to two years.
The major efficacy outcome measures were objective response rate (ORR) and duration of response (DOR) as assessed by blinded independent central radiologists' (BICR) review according to response evaluation criteria in solid tumours (RECIST) v1.1. The efficacy population was defined as patients who had measurable disease by BICR at baseline and had minimum of 24 weeks follow-up or had less than 24 weeks of follow-up and discontinued due to adverse events or disease progression.
A total of 108 patients with dMMR/MSI-H EC were evaluated for efficacy in the GARNET study.
Among these 108 patients, the baseline characteristics were: median age of 64 years (50.0% aged 65 years or older); 77.8% White, 4.6% Asian, 1.9% Black; and eastern cooperative oncology group (ECOG) performance status (PS) 0 (38.9%) or 1 (61.1%). At the time of diagnosis, 18.5% of the patients with dMMR/MSI-H EC were International Federation of Gynecology and Obstetrics (FIGO) Stage IV. At study entry (the most recent FIGO stage), 65.7% of the patients were FIGO Stage IV. The median number of prior therapies for recurrent or advanced EC was one and all had received treatment with a platinum-containing regimen. Thirty-six percent of patients received two or more prior lines of therapy.
The identification of dMMR/MSI-H tumour status was prospectively determined based on local testing.
Local diagnostic assays (IHC, PCR or NGS) available at the sites were used for the detection of the dMMR/MSI-H expression in tumour material. Most of the sites used IHC as it was the most common assay available.
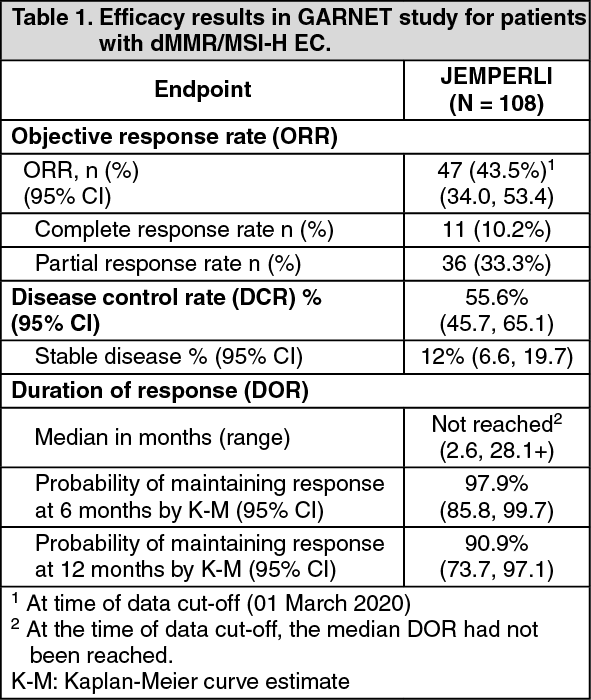
Table 1 includes the efficacy data for the 108 patients (median follow-up of 16.3 months). The overall median treatment duration was 26.0 weeks. Twelve patients (9.3%) received treatment for a duration ≥96 weeks (22 months).
Of the 108 patients, 78.3% of responders had an ongoing response of 6 months or longer.
Efficacy results are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEfficacy and PD-L1 status: Clinical activity was observed regardless of tumour PD-L1 combined positive score (CPS) by IHC. The relationship between PD-L1 status and efficacy was analysed post-hoc in patients with available tissue samples (N = 80) among the efficacy population from Cohort A1 using a data cut-off date of 01 March 2020. Among 23 patients with PD-L1 CPS <1%, ORR was 30.4% (7/23, 95% CI 13.2, 52.9) and among 58 patients with PD-L1 CPS ≥1%, ORR was 55.2% (32/58, 95% CI 41.5, 68.3).
Elderly patients: Of the 108 patients treated with dostarlimab in the efficacy population, 50.0% were older than 65 years.
Consistent results were observed in the elderly population, where the ORR by BICR (95% CI) was 42.6% (29.2%, 56.8%) in patients ≥65 years.
Pharmacokinetics: Dostarlimab was characterised using population PK analysis from 546 patients with various solid tumours, including 150 patients with EC. When dosed at the recommended therapeutic dose (500 mg administered intravenously every 3 weeks for 4 doses, followed by 1,000 mg every 6 weeks), dostarlimab shows an approximate two-fold accumulation (Cmin) starting cycle 4 through cycle 12, consistent with the terminal half-life (t1/2).
Absorption: Dostarlimab is administered via the intravenous route and therefore estimates of absorption are not applicable.
Distribution: The mean volume of distribution of dostarlimab at steady state is approximately 5.3 L (CV% of 12.3%).
Biotransformation: Dostarlimab is a therapeutic mAb IgG4 that is expected to be catabolised into small peptides, amino acids, and small carbohydrates by lysosome through fluid-phase or receptor-mediated endocytosis. The degradation products are eliminated by renal excretion or returned to the nutrient pool without biological effects.
Elimination: The mean clearance is 0.007 L/h (CV% of 31.3%) at steady state. The t1/2 at steady state is 25.4 days (CV% of 24.0%).
Linearity/non-linearity: Exposure (both maximum concentration [Cmax] and the area under the concentration-time curve, [AUC0-tau] and [AUC0-inf]) was approximately dose proportional.
Pharmacokinetic/pharmacodynamic relationship: Based on exposure efficacy and safety relationships, there are no clinically significant differences in efficacy and safety when doubling the exposure of dostarlimab. Full receptor occupancy as measured by both the direct PD-1 binding and interleukin 2 (IL-2) production functional assay was maintained throughout the dosing interval at the recommended therapeutic dosing regimen.
Special populations: A population PK analysis of the patient data indicates that there are no clinically important effects of age (range: 24 to 86 years), gender or race, ethnicity, or tumour type on the clearance of dostarlimab.
Renal impairment: Renal impairment was evaluated based on the estimated creatinine clearance [CLCR mL/min] (normal: CLCR ≥90 mL/min, n = 173; mild: CLCR = 60-89 mL/min, n = 210; moderate: CLCR = 30-59 mL/min, n = 90; severe: CLCR = 15-29 mL/min, n = 3 and ESRD: CLCR <15 mL/min, n = 1). The effect of renal impairment on the clearance of dostarlimab was evaluated by population pharmacokinetic analyses in patients with mild or moderate renal impairment compared to patients with normal renal function. No clinically important differences in the clearance of dostarlimab were found between patients with mild or moderate renal impairment and patients with normal renal function. There are limited data in patients with severe renal impairment.
Hepatic impairment: Hepatic impairment was evaluated as defined using the US National Cancer Institute criteria of hepatic dysfunction by total bilirubin and AST (Normal: total bilirubin (TB) & AST < = upper limit of normal (ULN), n = 425; mild: TB > ULN to 1.5 ULN or AST > ULN, n = 48; and moderate: TB > 1.5-3 ULN, any AST, n = 4). The effect of hepatic impairment on the clearance of dostarlimab was evaluated by population pharmacokinetic analyses in patients with mild hepatic impairment compared to patients with normal hepatic function. No clinically important differences in the clearance of dostarlimab were found between patients with mild hepatic impairment and normal hepatic function. There are limited data in patients with moderate hepatic impairment and no data in patients with severe hepatic impairment.
Toxicology: Preclinical safety data: Nonclinical data reveal no special hazard for humans based on repeat-dose toxicity studies of duration up to 3 months in the cynomolgus monkey. No studies have been performed to assess the potential of dostarlimab for carcinogenicity or genotoxicity. Animal reproduction and development toxicity studies have not been conducted with dostarlimab. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the foetus and to result in an increase in foetal loss. These results indicate a potential risk that administration of dostarlimab during pregnancy could cause foetal harm, including increased rates of abortion or stillbirth.
No notable effects on the male and female reproductive organs were observed in monkeys in the 1-month and 3-month repeat-dose toxicology studies; however, these results may not be representative at all of the potential clinical risk because of the immaturity of the reproductive system of animals used in the studies. Therefore, fertility toxicity remains unknown.