


 Sign Out
Sign Out

The most commonly reported events are bleedings occurring in approximately 14 % of patients; the frequency of major bleeds (including wound site bleedings) is less than 2 %.
Although rare in frequency in clinical trials, major or severe bleeding may occur and, regardless of location, may lead to disabling, life-threatening or even fatal outcomes.
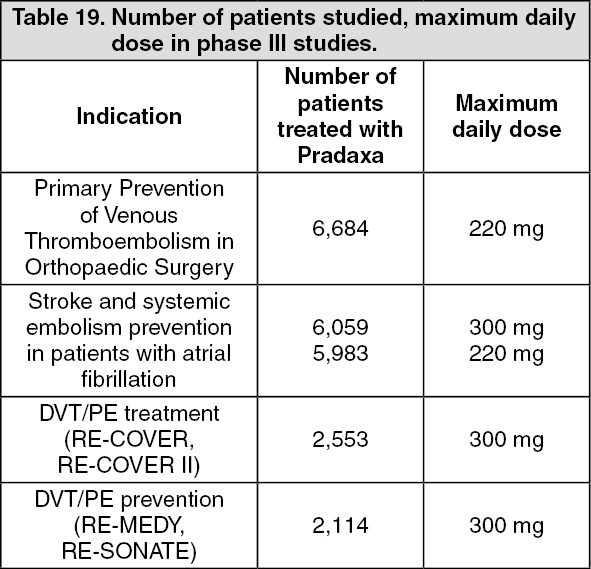
Summary of the safety profile for 110mg: The safety of Pradaxa has been evaluated in ten phase III studies including 23,393 patients exposed to Pradaxa (see Table 19).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn total, about 9 % of patients treated for elective hip or knee surgery (short-term treatment for up to 42 days), 22 % of patient with atrial fibrillation treated for the prevention of stroke and systemic embolism (long-term treatment for up to 3 years), 14 % of patient treated for DVT/PE and 15 % of patients treated for DVT/PE prevention experienced adverse reactions.
The most commonly reported events are bleedings occurring in approximately 14 % of patients treated short-term for elective hip or knee replacement surgery, 16.6 % in patients with atrial fibrillation treated long-term for the prevention of stroke and systemic embolism, and in 14.4 % of patients treated for DVT/PE. Furthermore, bleeding occurred in 19.4% of patients in the DVT/PE prevention trial RE-MEDY and in 10.5% of patient in the DVT/PE prevention trial RE-SONATE.
Since the patient populations treated in the three indications are not comparable and bleeding events are distributed over several System Organ Classes (SOC), a summary description of major and any bleeding are broken down by indication and given in Tables 22-26 as follows.
Although low in frequency in clinical trials, major or severe bleeding may occur and, regardless of location, may lead to disabling, life-threatening or even fatal outcomes.
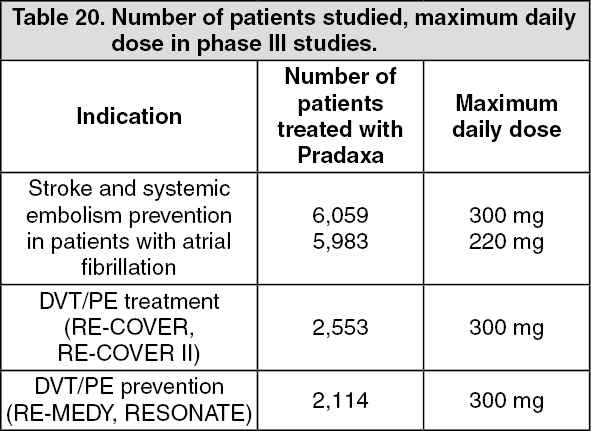
Summary of the safety profile for 150mg: The safety of Pradaxa has been evaluated in a pivotal study investigating the prevention of stroke and systemic embolism in patients with atrial fibrillation, in two active controlled DVT/PE treatment trials and in one active controlled DVT/PE prevention trial. In these four phase III trials, 16,709 patients were exposed to Pradaxa (see Table 20).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn total, 22 % of patient with atrial fibrillation treated for the prevention of stroke and systemic embolism (long-term treatment for up to 3 years), 14 % of patients treated for DVT/PE and 15 % of patients treated for DVT/PE prevention experienced adverse reactions.
The most commonly reported events are bleedings occurring in approximately 16.6 % in patients with atrial fibrillation treated long-term for the prevention of stroke and systemic embolism and in 14.4 % of patients treated for DVT/PE. Furthermore, bleeding occurred in 19.4 % of patients in the DVT/PE prevention trial RE-MEDY and in 10.5 % of patients in the DVT/PE trial RE-SONATE.
Since the patient population treated in the three indications are not comparable and bleeding events are distributed over several System Organ Classes (SOC), a summary description of major and any bleeding are broken down by indication and are provided in Tables 23-26 as follows.
Although low in frequency in clinical trials, major or severe bleeding may occur and, regardless of location, may lead to disabling, life-threatening or even fatal outcomes.
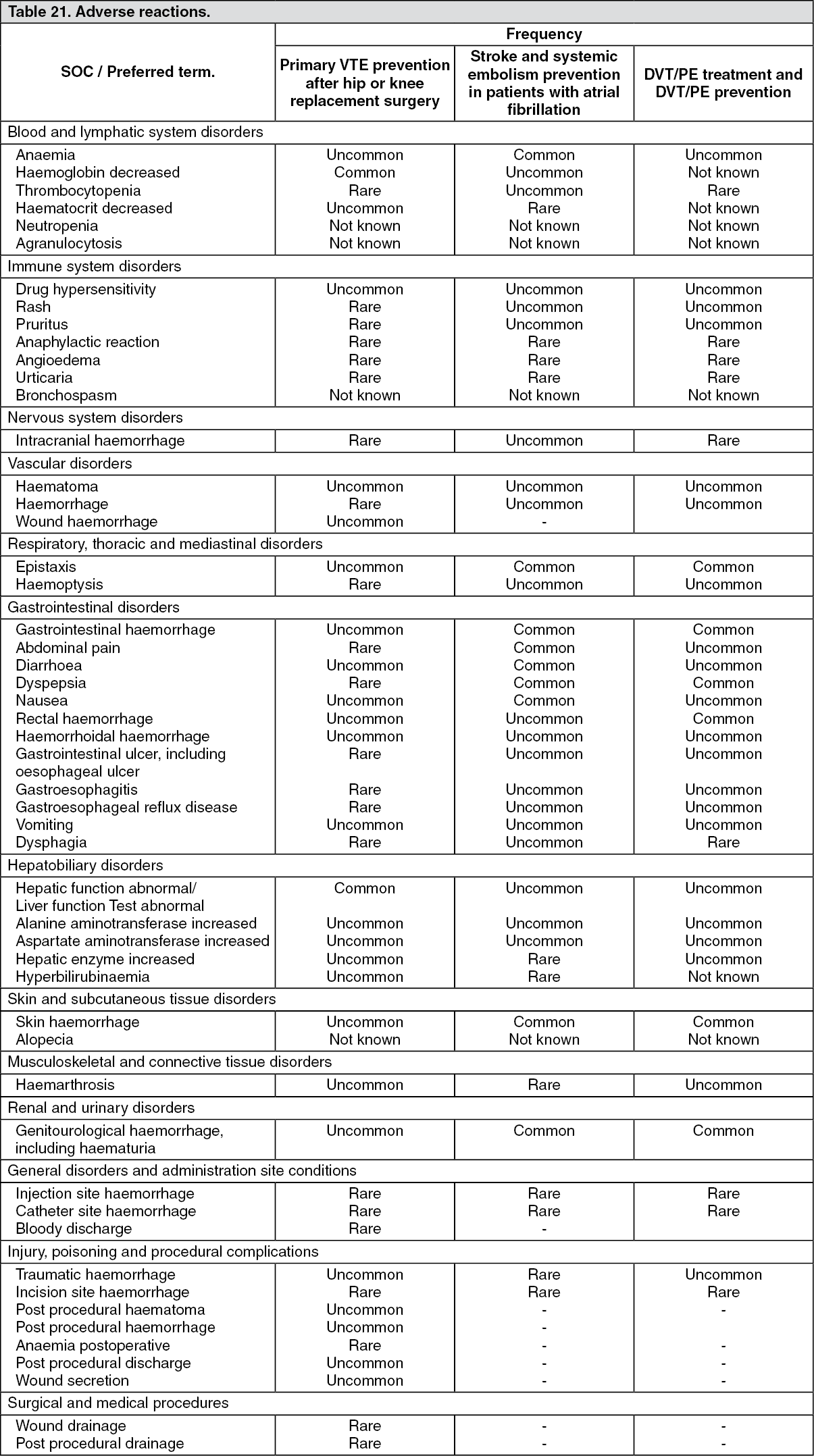
Tabulated list of adverse reactions: Table 21 shows the adverse reactions identified from studies and post-marketing data in the indications primary VTE prevention after hip or knee replacement surgery, prevention of thromboembolic stroke and systemic embolism in patients with atrial fibrillation, DVT/PE treatment and in DVT/PE prevention. They are ranked under headings of System Organ Class (SOC) and frequency using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), not known (cannot be estimated from the available data). (See Table 21.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Bleeding reactions: Due to the pharmacological mode of action, the use of Pradaxa may be associated with an increased risk of occult or overt bleeding from any tissue or organ. The signs, symptoms, and severity (including fatal outcome) will vary according to the location and degree or extent of the bleeding and/or anaemia. In the clinical studies mucosal bleedings (e.g. gastrointestinal, genitourinary) were seen more frequently during long term Pradaxa treatment compared with VKA treatment. Thus, in addition to adequate clinical surveillance, laboratory testing of haemoglobin/haematocrit is of value to detect occult bleeding. The risk of bleedings may be increased in certain patient groups e.g. those patients with moderate renal impairment and/or on concomitant treatment affecting haemostasis or strong P-gp inhibitors (see Haemorrhagic risk under Precautions). Haemorrhagic complications may present as weakness, paleness, dizziness, headache or unexplained swelling, dyspnoea, and unexplained shock.
Known bleeding complications such as compartment syndrome and acute renal failure due to hypoperfusion have been reported for Pradaxa. Therefore, the possibility of haemorrhage is to be considered in evaluating the condition in any anticoagulated patient. A specific reversal agent for dabigatran, idarucizumab, is available in case of uncontrollable bleeding (see Overdosage).
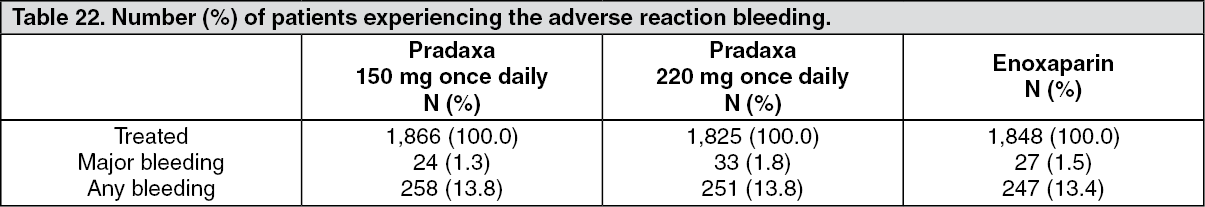
Primary Prevention of Venous Thromboembolism in Orthopaedic Surgery: Table 22 shows the number (%) of patients experiencing the adverse reaction bleeding during the treatment period in the VTE prevention in the two pivotal clinical trials, according to dose. (See Table 22.)
 Click on icon to see table/diagram/image
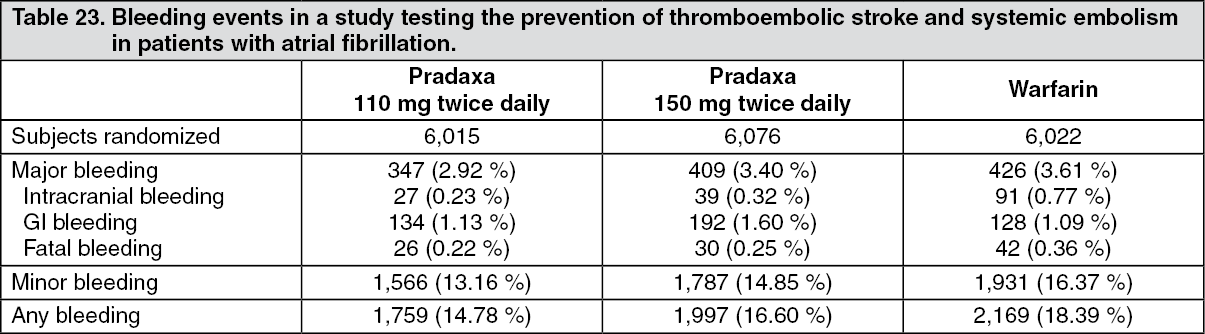
Click on icon to see table/diagram/imagePrevention of stroke and systemic embolism in adult patients with NVAF with one or more risk factors: Table 23 shows bleeding events broken down to major and any bleeding in the pivotal study testing the prevention of thromboembolic stroke and systemic embolism in patients with atrial fibrillation. (See Table 23.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSubjects randomized to Pradaxa 110 mg twice daily or 150 mg twice daily had a significantly lower risk for life-threatening bleeds and intracranial bleeding compared to warfarin [p < 0.05]. Both dose strengths of Pradaxa had also a statistically significant lower total bleed rate. Subjects randomized to 110 mg Pradaxa twice daily had a significantly lower risk for major bleeds compared with warfarin (hazard ratio 0.81 [p=0.0027]). Subjects randomized to 150 mg Pradaxa twice daily had a significantly higher risk for major GI bleeds compared with warfarin (hazard ratio 1.48 [p=0.0005]). This effect was seen primarily in patients ≥ 75 years.
The clinical benefit of dabigatran with regard to stroke and systemic embolism prevention and decreased risk of ICH compared to warfarin is preserved across individual subgroups, e.g. renal impairment, age, concomitant medicinal product use such as anti-platelets or P-gp inhibitors. While certain patient subgroups are at an increased risk of major bleeding when treated with an anticoagulant, the excess bleeding risk for dabigatran is due to GI bleeding, typically seen within the first 3-6 months following initiation of Pradaxa therapy.
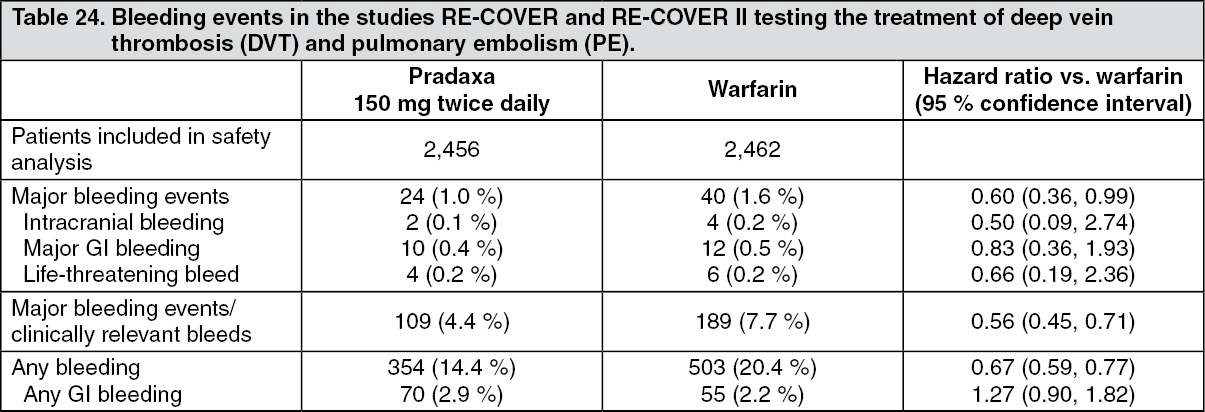
Treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and prevention of recurrent DVT and PE in adults (DVT/PE treatment): Table 24 shows bleeding events in the pooled pivotal studies RE-COVER and RE-COVER II testing the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE). In the pooled studies the primary safety endpoints of major bleeding, major or clinically relevant bleeding and any bleeding were significantly lower than warfarin at a nominal alpha level of 5 %. (See Table 24.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageBleeding events for both treatments are counted from the first intake of Pradaxa or warfarin after the parenteral therapy has been discontinued (oral only treatment period). This includes all bleeding events, which occurred during Pradaxa therapy. All bleeding events which occurred during warfarin therapy are included except for those during the overlap period between warfarin and parenteral therapy.
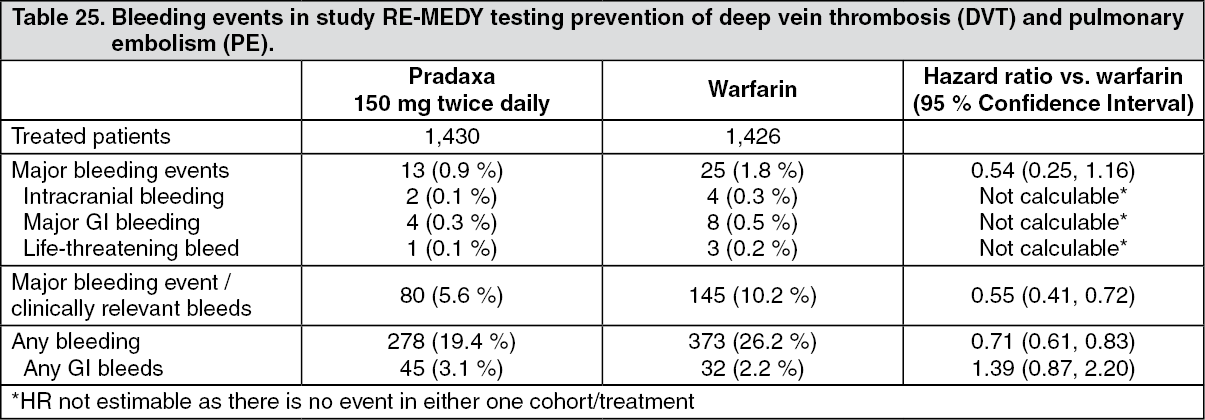
Table 25 shows bleeding events in pivotal study RE-MEDY testing prevention of deep vein thrombosis (DVT) and pulmonary embolism (PE). Some bleeding events (MBEs/CRBEs; any bleeding) were significantly lower at a nominal alpha level of 5% in patients receiving Pradaxa as compared with those receiving warfarin. (See Table 25.)
 Click on icon to see table/diagram/image
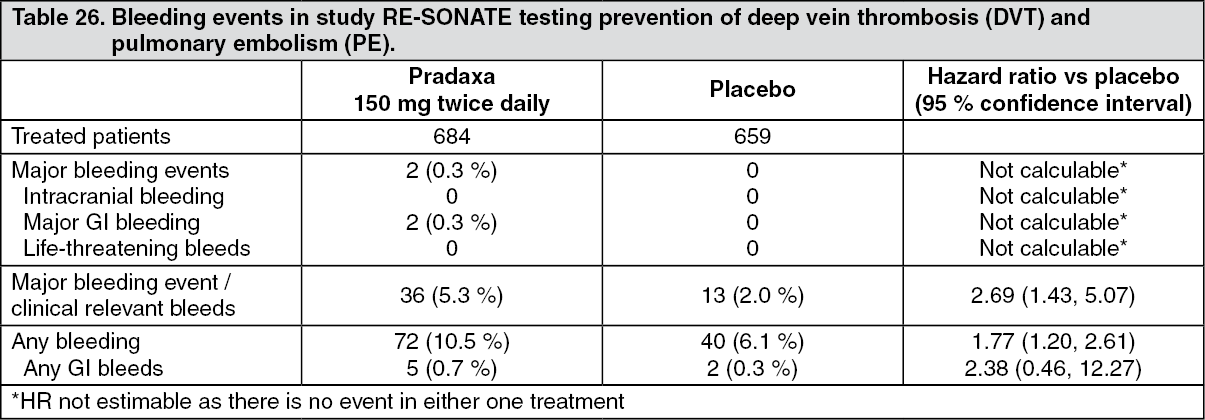
Click on icon to see table/diagram/imageTable 26 shows bleeding events in pivotal study RE-SONATE testing prevention of deep vein thrombosis (DVT) and pulmonary embolism (PE). The rate of the combination of MBEs/CRBEs and the rate of any bleeding was significantly lower at a nominal alpha level of 5 % in patients receiving placebo as compared with those receiving Pradaxa. (See Table 26.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAgranulocytosis and neutropenia: Agranulocytosis and neutropenia have been reported very rarely during post approval use of Pradaxa. Because adverse reactions are reported in the postmarketing surveillance setting from a population of uncertain size, it is not possible to reliably determine their frequency. The reporting rate was estimated as 7 events per 1 million patient years for agranulocytosis and as 5 events per 1 million patient years for neutropenia.
Paediatric population (DVT/PE): In the clinical study 1160.88 in total, 9 adolescent patients (age 12 to < 18 years) with diagnosis of primary VTE received an initial oral dose of dabigatran etexilate of 1.71 (± 10 %) mg/kg bodyweight. Based on dabigatran concentrations as determined by the diluted thrombin time test and clinical assessment, the dose was adjusted to the target dose of 2.14 (± 10%) mg/kg bodyweight of dabigatran etexilate. On treatment 2 (22.1 %) patients experienced mild related adverse events (gastrooesophageal reflux / abdominal pain; abdominal discomfort) and 1 (11.1 %) patient experienced a not related serious adverse event (recurrent VTE of the leg) in the post treatment period > 3 days after stop of dabigatran etexilate.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions.
View ADR Monitoring Form