


 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Mechanism of action: Bedaquiline is a diarylquinoline. Bedaquiline specifically inhibits mycobacterial ATP (adenosine 5'-triphosphate) synthase, an essential enzyme for the generation of energy in Mycobacterium tuberculosis. The inhibition of ATP synthase leads to bactericidal effects for both replicating and non-replicating tubercle bacilli.
Pharmacodynamic effects: Bedaquiline has activity against Mycobacterium tuberculosis with a minimal inhibitory concentration (MIC) for drug-sensitive as well as drug-resistant strains (multi-drug resistant including pre-extensively drug resistant strains, extensively drug resistant strains) in the range of ≤ 0.008-0.12 mg/l. The N-monodesmethyl metabolite (M2) is not thought to contribute significantly to clinical efficacy given its lower average exposure (23% to 31%) in humans and lower antimycobacterial activity (3- to 6-fold lower) compared to the parent compound.
The intracellular bactericidal activity of bedaquiline in primary peritoneal macrophages and in a macrophage-like cell line was greater than its extracellular activity. Bedaquiline is also bactericidal against dormant (non-replicating) tubercle bacilli. In the mouse model for TB infection, bedaquiline has demonstrated bactericidal and sterilizing activities.
Bedaquiline is bacteriostatic for many non-tuberculous mycobacterial species. Mycobacterium xenopi, Mycobacterium novocastrense, Mycobacterium shimoidei and non-mycobacterial species are considered inherently resistant to bedaquiline.
Pharmacokinetic/pharmacodynamic relationship: Within the concentration range achieved with the therapeutic dose, no pharmacokinetic/pharmacodynamic relationship was observed in patients.
Mechanisms of resistance: Acquired resistance mechanisms that affect bedaquiline MICs include mutations in the atpE gene, which codes for the ATP synthase target, and in the Rv0678 gene, which regulates the expression of the MmpS5-MmpL5 efflux pump. Target-based mutations generated in preclinical studies lead to 8- to 133-fold increases in bedaquiline MIC, resulting in MICs ranging from 0.25 to 4.0 mg/l. Efflux-based mutations have been seen in preclinical and clinical isolates. These lead to 2- to 8-fold increases in bedaquiline MICs, resulting in bedaquiline MICs ranging from 0.25 to 0.50 mg/l. Isolates with efflux-based mutations are also less susceptible to clofazimine.
However no clear relationship between increased post-baseline bedaquiline MICs and microbiologic outcomes was observed in the phase 2 trials where bedaquiline was given for 24 weeks, followed by continuation of the background regimen.
Susceptibility testing breakpoints: When available, the clinical microbiology laboratory should provide the physician with the results of in vitro susceptibility test results for antimicrobial medicinal products used in resident hospitals as periodic reports that describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting a combination of antibacterial medicinal products for treatment.
Breakpoints: Minimal inhibitory concentration (MIC) breakpoints are as follows: Epidemiological Cut-Off (ECOFF) 0.25 mg/l; Clinical Breakpoints S ≤ 0.25 mg/l; R > 0.25 mg/l; S = susceptible; R = resistant.
Commonly susceptible species: Mycobacterium tuberculosis.
Inherently resistant organisms: Mycobacterium xenopi; Mycobacterium novocastrense; Mycobacterium shimoidei; Non-mycobacterial species.
Clinical efficacy and safety: The following definitions applies for resistance categories used: Multi-drug resistant Mycobacterium tuberculosis (MDRH&R-TB): isolate resistant to at least isoniazid and rifampicin, but susceptible to fluoroquinolones and second line injectable agents.
Pre-extensively drug resistant tuberculosis (pre-XDR-TB): isolate resistant to isoniazid, rifampicin, and either any fluoroquinolone or at least one second line injectable agent (but not to both a fluoroquinolone and a second line injectable agent).
Extensively drug resistant tuberculosis (XDR-TB): isolate resistant to isoniazid, rifampicin, any fluoroquinolone, and at least one second line injectable agent.
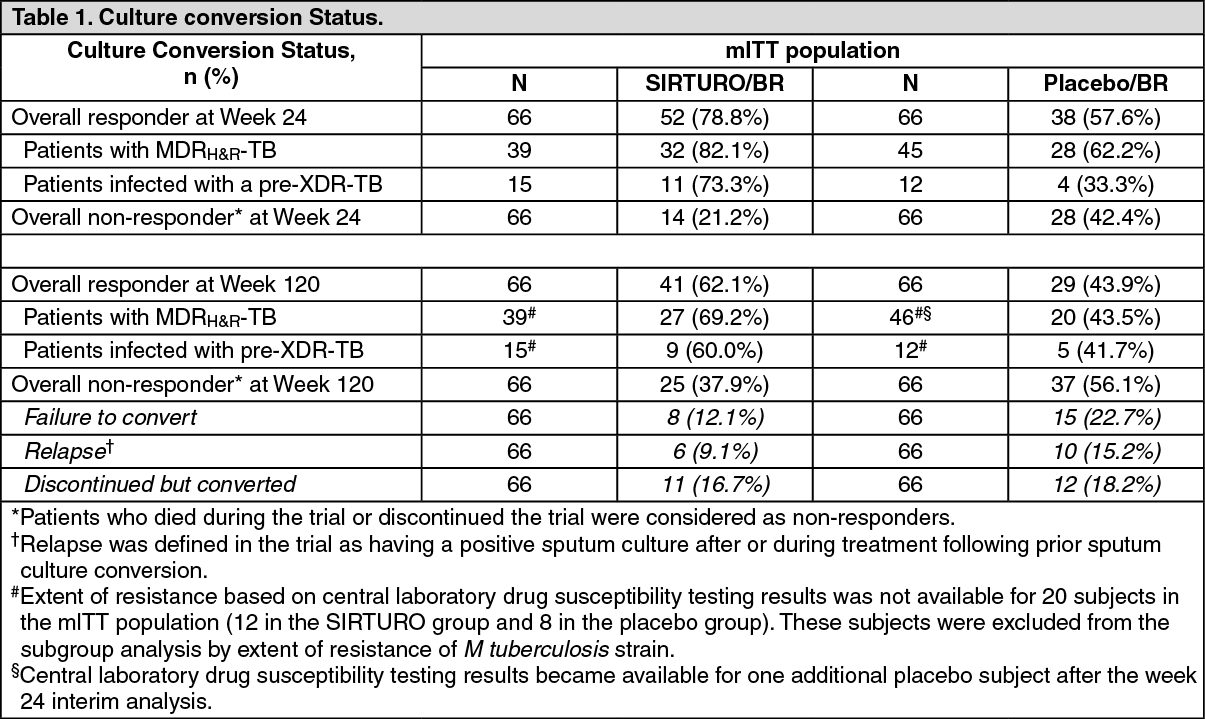
A Phase IIb, placebo-controlled, double-blind, randomised trial (C208) evaluated the antibacterial activity, safety, and tolerability of SIRTURO in newly diagnosed patients with sputum smear-positive pulmonary MDRH&R- and pre-XDR-TB. Patients received SIRTURO (n = 79) or placebo (n = 81) for 24 weeks, both in combination with a preferred 5-drug background regimen (BR) consisting of ethionamide, kanamycin, pyrazinamide, ofloxacin, and cycloserine/terizidone. After the 24-week investigational period, the background regimen was continued to complete 18 to 24 months of total multi-drug resistant Mycobacterium tuberculosis treatment. A final evaluation was conducted at Week 120. Main demographics were as follows: 63.1% were males, median age 34 years, 35% were Black, and 15% were HIV positive. Cavitation in one lung was seen in 58% of patients, and in both lungs in 16%. For patients with full characterisation of resistance status, 76% (84/111) were infected with an MDRH&R-TB strain and 24% (27/111) with a pre-XDR-TB strain.
SIRTURO was administered as 400 mg once daily for the first 2 weeks, and as 200 mg 3 times/week for the following 22 weeks.
The primary outcome parameter was the time to sputum culture conversion (i.e. the interval between the first SIRTURO intake and the first of two consecutive negative liquid cultures from sputum collected at least 25 days apart) during treatment with SIRTURO or placebo (median time to conversion was 83 days for the SIRTURO group, 125 days for the placebo group (hazard ratio, 95% CI: 2.44 [1.57; 3.80]), p < 0.0001).
In the SIRTURO group, no or only minor differences in time to culture conversion and culture conversion rates were observed between patients with pre-XDR-TB and patients with MDRH&R-TB.
Response rates at week 24 and week 120 (i.e. around 6 months after stopping all therapy) are presented in table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageStudy C209 evaluated the safety, tolerability, and efficacy of 24 weeks treatment with open-label SIRTURO as part of an individualized treatment regimen in 233 patients who were sputum smear positive within 6 months prior to screening. This study included patients of all three resistance categories (MDRH&R-, pre-XDR- and XDR-TB).
The primary efficacy endpoint was the time to sputum culture conversion during treatment with SIRTURO (median 57 days, for 205 patients with sufficient data). At week 24, sputum culture conversion was seen in 163/205 (79.5%) patients. Conversion rates at week 24 were highest (87.1%; 81/93) in patients with MDRH&R-TB, 77.3% (34/44) in pre-XDR-TB patients and lowest (54.1%; 20/37) in XDR-TB patients. Extent of resistance based on central laboratory drug susceptibility testing results was not available for 32 subjects in the mITT population. These subjects were excluded from the subgroup analysis by extent of resistance of Mycobacterium tuberculosis strain.
At week 120, sputum culture conversion was seen in 148/205 (72.2%) patients. Conversion rates at week 120 were highest (73.1%; 68/93) in patients with MDRH&R-TB, 70.5% (31/44) in pre-XDR-TB patients and lowest (62.2%; 23/37) in XDR-TB patients.
At both week 24 and week 120, responder rates were higher for patients on 3 or more active substances (in vitro) in their background regimen.
Of the 163 patients who were responders at week 24, 139 patients (85.3%) were still responders at week 120. Twenty-four of these 24-week responders (14.7%) were considered non-responders at week 120, of which 19 patients had prematurely discontinued the trial while being culture converted and 5 patients had experienced relapse. Of the 42 patients who were non-responders at week 24, confirmed culture conversion after week 24 (i.e., after bedaquiline dosing ended but the background regimen was continued) occurred in 9 patients (21.4%) and was maintained at week 120.
Pharmacokinetics: The pharmacokinetic properties of bedaquiline have been evaluated in adult healthy subjects and in adult multi-drug resistant tuberculosis-infected patients. Exposure to bedaquiline was lower in multi-drug resistant tuberculosis-infected patients than in healthy subjects.
Absorption: Maximum plasma concentrations (Cmax) are typically achieved at about 5 hours post-dose. Cmax and the area under the plasma concentration-time curve (AUC) increased proportionally up to the highest doses studied (700 mg single-dose and once daily 400 mg multiple doses). Administration of bedaquiline with food increased the relative bioavailability by about 2-fold compared to administration under fasted conditions. Therefore, bedaquiline should be taken with food to enhance its oral bioavailability.
Distribution: The plasma protein binding of bedaquiline is > 99.9% in all species tested, including human. The plasma protein binding of the N-monodesmethyl metabolite (M2) in humans is at least 99.8%. In animals, bedaquiline and its active N-monodesmethyl metabolite (M2) are extensively distributed to most tissues, however, brain uptake was low.
Biotransformation: CYP3A4 was the major CYP isoenzyme involved in vitro in the metabolism of bedaquiline and the formation of the N-monodesmethyl metabolite (M2).
In vitro, bedaquiline does not significantly inhibit the activity of any of the CYP450 enzymes tested (CYP1A2, CYP2A6, CYP2C8/9/10, CYP2C19, CYP2D6, CYP2E1, CYP3A4, CYP3A4/5 and CYP4A) and does not induce CYP1A2, CYP2C9 or CYP2C19 activities.
Bedaquiline and M2 were not substrates of P-gp in vitro. Bedaquiline was a weak OCT1, OATP1B1 and OATP1B3 substrate in vitro, while M2 was not. Bedaquiline was not a substrate of MRP2 and BCRP in vitro. Bedaquiline and M2 did not inhibit the transporters P-gp, OATP1B1, OATP1B3, BCRP, OAT1, OAT3, OCT1, OCT2, MATE1 and MATE2 at clinically relevant concentrations in vitro. An in vitro study indicated a potential for bedaquiline to inhibit BCRP at the concentrations achieved in the intestine after oral administration. The clinical relevance is unknown.
Elimination: Based on the preclinical studies, the bulk of the administered dose is eliminated in faeces. The urinary excretion of unchanged bedaquiline was < 0.001% of the dose in clinical studies, indicating that renal clearance of unchanged active substance is insignificant. After reaching Cmax, bedaquiline concentrations decline tri-exponentially. The mean terminal
elimination half-life of both bedaquiline and the active N-monodesmethyl metabolite (M2) is about 5 months (ranging from 2 to 8 months). This long terminal elimination phase likely reflects slow release of bedaquiline and M2 from peripheral tissues.
Special populations: Hepatic impairment: A single-dose study of SIRTURO in 8 subjects with moderate hepatic impairment (Child-Pugh B) demonstrated exposure to bedaquiline and M2 (AUC672h) was 19% lower compared to healthy subjects. No dose adjustment is deemed necessary in patients with mild or moderate hepatic impairment. Bedaquiline has not been studied in patients with severe hepatic impairment (see Dosage & Administration).
Renal impairment: SIRTURO has mainly been studied in patients with normal renal function. Renal excretion of unchanged bedaquiline is insignificant (< 0.001%).
In a population pharmacokinetic analysis of tuberculosis patients treated with SIRTURO 200 mg three times a week, creatinine clearance (range: 40 to 227 ml/min) was not found to influence the pharmacokinetic parameters of bedaquiline. It is therefore not expected that mild or moderate renal impairment will have a clinically relevant effect on the exposure to bedaquiline. However, in patients with severe renal impairment (creatinine clearance < 30 ml/min) or end-stage renal disease requiring haemodialysis or peritoneal dialysis, bedaquiline concentrations may be increased due to alteration of active substance absorption, distribution, and metabolism secondary to renal dysfunction. As bedaquiline is highly bound to plasma proteins, it is unlikely that it will be significantly removed from plasma by haemodialysis or peritoneal dialysis.
Paediatric patients: The pharmacokinetics of SIRTURO in paediatric patients have not been evaluated.
Elderly patients: There is limited clinical data (n = 2) on the use of SIRTURO in tuberculosis patients aged 65 years and older.
In a population pharmacokinetic analysis of tuberculosis patients (age range 18 years to 68 years) treated with SIRTURO age was not found to influence the pharmacokinetics of bedaquiline.
Race: In a population pharmacokinetic analysis of tuberculosis patients treated with SIRTURO, exposure to bedaquiline was found to be lower in Black patients than in patients from other race categories. This low exposure was not considered to be clinically relevant as no clear relationship between exposure to bedaquiline and response has been observed in clinical trials. Furthermore, response rates in patients that completed the bedaquiline treatment period were comparable between different race categories in the clinical trials.
Gender: In a population pharmacokinetic analysis of tuberculosis patients treated with SIRTURO no clinically relevant difference in exposure between men and women were observed.
Toxicology: Preclinical safety data: Animal toxicology studies have been conducted with bedaquiline administration up to 3 months in mice, up to 6 months in rats, and up to 9 months in dogs. The plasma bedaquiline exposure (AUC) in rats and dogs was similar to that observed in humans. Bedaquiline was associated with effects in target organs which included monocytic phagocytic system (MPS), skeletal muscle, liver, stomach, pancreas and heart muscle.
All of these toxicities except effects on MPS were monitored clinically. In the MPS of all species, pigment-laden and/or foamy macrophages were also seen in various tissues, consistent with phospholipidosis. The significance of phospholipidosis in humans is unknown. Most of the observed changes occurred after prolonged daily dosing and subsequent increases in plasma and tissue concentrations of the active substance. After treatment cessation, all indications of toxicity exhibited at least partial recovery to good recovery.
In a rat carcinogenicity study, bedaquiline, at the high doses of 20 mg/kg/day in males and 10 mg/kg/day in females, did not induce any treatment-related increases in tumour incidences. Compared to the exposures (AUC) observed in subjects with MDR-TB in the bedaquiline phase II trials, the exposures (AUC) in rats at high doses were similar in males and 2-fold higher in females for bedaquiline, and 3-fold higher in males and 2-fold higher in females for M2.
In vitro and in vivo genotoxicity tests indicated that bedaquiline did not have any mutagenic or clastogenic effects.
Bedaquiline had no effects on fertility when evaluated in female rats. Three of 24 male rats treated with high bedaquiline doses failed to produce offspring in the fertility study. Normal spermatogenesis and a normal amount of spermatozoa in the epidydimides were noted in these animals. No structural abnormalities in the testes and epididymides were seen after up to 6-months of bedaquiline treatment. No relevant bedaquiline-related effects on developmental toxicity parameters were observed in rats and rabbits. The corresponding plasma exposure (AUC) was 2-fold higher in rats compared to humans. In the rat, no adverse effects were observed in a pre- and post-natal development study at maternal plasma exposure (AUC) similar to humans and exposure in the offspring 3-fold higher than in adult humans. There was no effect of maternal treatment with bedaquiline at any dose level on sexual maturation, behavioural development, mating performance, fertility or reproductive capacity of the F1 generation animals. Body weight decreases in pups were noted in high dose groups during the lactation period after exposure to bedaquiline via milk and were not a consequence of in utero exposure. Concentrations of bedaquiline in milk were 6- to 12-fold higher that the maximum concentration observed in maternal plasma.
Environmental Risk Assessment (ERA): Environmental risk assessment studies have shown that bedaquiline has the potential to be persistent, bioaccumulative and toxic to the environment (see Cautions for Usage).