Pharmacotherapeutic group: Drugs for treatment of bone diseases, bisphosphonate.
ATC code: M05BA08.
Pharmacology: Pharmacodynamics: Zoledronic acid belongs to the class of bisphosphonates and acts primarily on bone. It is an inhibitor of osteoclastic bone resorption.
The selective action of bisphosphonates on bone is based on their high affinity for mineralised bone, but the precise molecular mechanism leading to the inhibition of osteoclastic activity is still unclear. In long-term animal studies, zoledronic acid inhibits bone resorption without adversely affecting the formation, mineralisation or mechanical properties of bone.
In addition to being a potent inhibitor of bone resorption, zoledronic acid also possesses several anti-tumour properties that could contribute to its overall efficacy in the treatment of metastatic bone disease. The following properties have been demonstrated in preclinical studies:
In vivo: Inhibition of osteoclastic bone resorption, which alters the bone marrow microenvironment making it less conducive to tumour cell growth, anti-angiogenic activity, anti-pain activity.
In vitro: Inhibition of osteoblast proliferation, direct cytostatic and pro-apoptotic activity on tumour cells, synergistic cytostatic effect with other anti-cancer drugs, anti-adhesion/invasion activity.
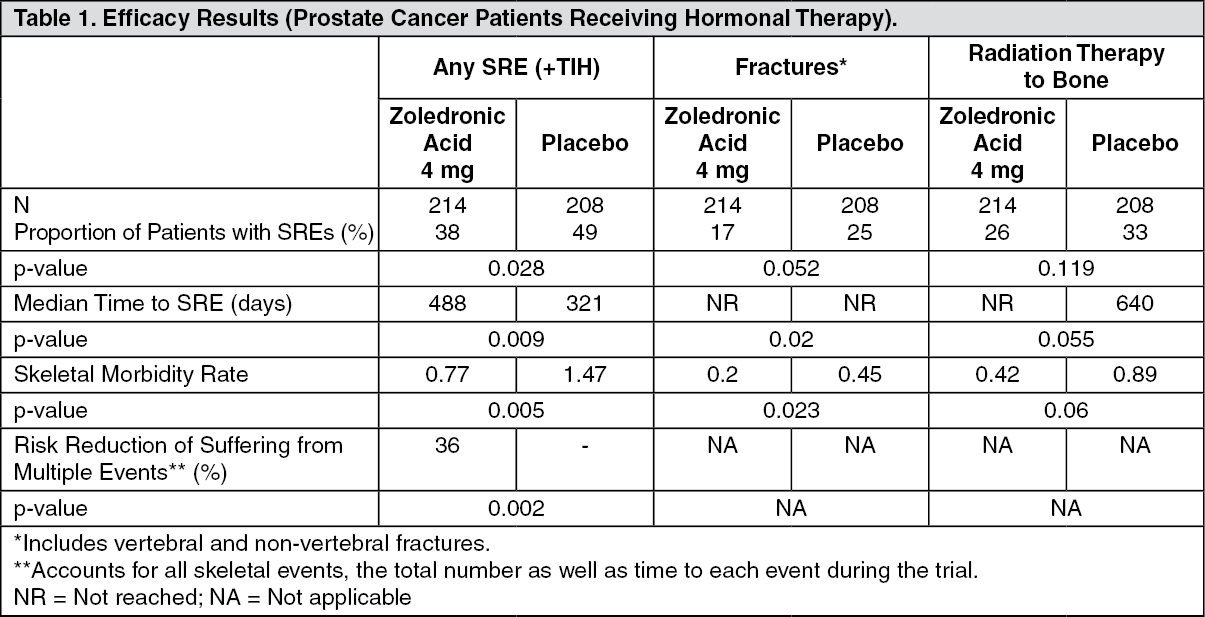
Clinical trial results in the prevention of skeletal related events in patients with advanced malignancies involving bone: The first randomised, double-blind, placebo-controlled study compared zoledronic acid 4 mg to placebo for the prevention of skeletal related events (SREs) in prostate cancer patients. Zoledronic acid 4 mg significantly reduced the proportion of patients experiencing at least one skeletal related event (SRE), delayed the median time to first SRE by >5 months and reduced the annual incidence of events per patient - skeletal morbidity rate. Multiple event analysis showed a 36% risk reduction in developing SREs in the zoledronic acid 4 mg group compared with placebo. Patients receiving zoledronic acid 4 mg reported less increase in pain than those receiving placebo and the difference reached significance at months 3, 9, 21 and 24. Fewer zoledronic acid 4 mg patients suffered pathological fractures. The treatment effects were less pronounced in patients with blastic lesions. Efficacy results are provided in Table 1. (See Table 1.)
In a second study including solid tumours other than breast or prostate cancer, zoledronic acid 4 mg significantly reduced the proportion of patients with an SRE, delayed the median time to first SRE by >2 months and reduced the skeletal morbidity rate. Multiple event analysis showed 30.7% risk reduction in developing SREs in the zoledronic acid 4 mg group compared with placebo. Efficacy results are provided in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In a third phase III randomised, double-blind trial, zoledronic acid 4 mg or 90 mg pamidronate every 3 to 4 weeks were compared in patients with multiple myeloma or breast cancer with at least one bone lesion. The results demonstrated that zoledronic acid 4 mg showed comparable efficacy to pamidronate 90 mg in the prevention of SREs. The multiple event analysis revealed a significant risk reduction of 16% in patients treated with zoledronic acid 4 mg in comparison with patients receiving pamidronate. Efficacy results are provided in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Zoledronic acid 4 mg was also studied in a double-blind, randomised, placebo-controlled trial in 228 patients with documented bone metastases from breast cancer to evaluate the effect of 4 mg zoledronic acid on the skeletal related event (SRE) rate ratio, calculated as the total number of SRE events (excluding hypercalcaemia and adjusted for prior fracture), divided by the total risk period. Patients received either 4 mg zoledronic acid or placebo every four weeks for one year. Patients were evenly distributed between zoledronic acid-treated and placebo groups.
The SRE rate (events/person year) was 0.628 for zoledronic acid and 1.096 for placebo. The proportion of patients with at least one SRE (excluding hypercalcaemia) was 29.8% in the zoledronic acid-treated group versus 49.6% in the placebo group (p=0.003). Median time to onset of the first SRE was not reached in the zoledronic acid-treated arm at the end of the study and was significantly prolonged compared to placebo (p=0.007). Zoledronic acid 4 mg reduced the risk of SREs by 41% in a multiple event analysis (risk ratio=0.59, p=0.019) compared with placebo.
In the zoledronic acid-treated group, statistically significant improvement in pain scores (using the Brief Pain Inventory, BPI) was seen 4 weeks and at every subsequent time point during the study, when compared to placebo (see Figure). The pain score for zoledronic acid was consistently below baseline and pain reduction was accompanied by a trend in reduced analgesics score. (See Figure.)
Click on icon to see table/diagram/image
Clinical trial results in the treatment of TIH: Clinical studies in tumour-induced hypercalacemia (TIH) demonstrated that the effect of zoledronic acid is characterised by decreases in serum calcium and urinary calcium excretion. In phase I dose finding studies in patients with mild to moderate tumour-induced hypercalcaemia (TIH), effective doses tested were in the range of approximately 1.2-2.5 mg.
To assess the effects of 4 mg zoledronic acid versus pamidronate 90 mg, the results of two pivotal multicentre studies in patients with TIH were combined in a pre-planned analysis. There was faster normalisation of corrected serum calcium at day 4 for 8 mg zoledronic acid and at day 7 for 4 mg zoledronic acid and 8 mg zolendronic acid. The following response rates were observed: (See Table 4).
Click on icon to see table/diagram/image
Median time to normocalcaemia was 4 days. Median time to relapse (re-increase of albumin-corrected serum calcium ≥2.9 mmol/L) was 30 to 40 days for patients treated with zoledronic acid versus 17 days for those treated with pamidronate 90 mg (p-values: 0.001 for 4 mg and 0.007 for 8 mg zoledronic acid). There were no statistically significant differences between the two zoledronic acid doses.
In clinical trials, 69 patients who relapsed or were refractory to initial treatment (zoledronic acid 4 mg, 8 mg or pamidronate 90 mg) were retreated with 8 mg zoledronic acid. The response rate in these patients was about 52%. Since those patients were retreated with the 8 mg dose only, there are no data available allowing comparison with the 4 mg zoledronic acid dose.
In clinical trials performed in patients with tumour-induced hypercalcaemia (TIH), the overall safety profile amongst all three treatment groups (zoledronic acid 4 and 8 mg and pamidronate 90 mg) was similar in types and severity.
Paediatric population: Clinical Trial Results in the Treatment of Severe Osteogenesis Imperfecta in Paediatric Patients Aged 1 to 17 years: The effects of intravenous zoledronic acid in the treatment of paediatric patients (age 1 to 17 years) with severe osteogenesis imperfecta (types I, III and IV) were compared to intravenous pamidronate in one international, multicentre, randomised, open-label study with 74 and 76 patients in each treatment group, respectively. The study treatment period was 12 months preceded by a 4- to 9-week screening period during which vitamin D and elemental calcium supplements were taken for at least 2 weeks. In the clinical programme, patients aged 1 to <3 years received 0.025 mg/kg zoledronic acid (up to a maximum single dose of 0.35 mg) every 3 months and patients aged 3 to 17 years received 0.05 mg/kg zoledronic acid (up to a maximum single dose of 0.83 mg) every 3 months. An extension study was conducted in order to examine the long-term general and renal safety of once yearly or twice yearly zoledronic acid over the 12-month extension treatment period in children who had completed one year of treatment with either zoledronic acid or pamidronate in the core study.
The primary endpoint of the study was the percent change from baseline in lumbar spine bone mineral density (BMD) after 12 months of treatment. Estimated treatment effects on BMD were similar, but the trial design was not sufficiently robust to establish non-inferior efficacy for zoledronic acid. In particular, there was no clear evidence of efficacy on incidence of fracture or on pain. Fracture adverse events of long bones in the lower extremities were reported in approximately 24% (femur) and 14% (tibia) of zoledronic acid-treated patients vs 12% and 5% of pamidronate-treated patients with severe osteogenesis imperfecta, regardless of disease type and causality but overall incidence of fractures was comparable for the zoledronic acid and pamidronate-treated patients: 43% (32/74) vs 41% (31/76). Interpretation of the risk of fracture is confounded by the fact that fractures are common events in patients with severe osteogenesis imperfecta as part of the disease process.
The type of adverse reactions observed in this population were similar to those previously seen in adults with advanced malignancies involving the bone (see Adverse Reactions). The adverse reactions ranked under headings of frequency, are presented in Table 5. The following conventional classification is used: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), not known (cannot be estimated from the available data). (See Table 5).
Click on icon to see table/diagram/image
In paediatric patients with severe osteogenesis imperfecta, zoledronic acid seems to be associated with more pronounced risks for acute phase reaction, hypocalcaemia and unexplained tachycardia, in comparison to pamidronate, but this difference declined after subsequent infusions.
The European Medicines Agency has waived the obligation to submit the results of studies with zoledronic acid in all subsets of the paediatric population in the treatment of tumour-induced hypercalcaemia and prevention of skeletal-related events in patients with advanced malignancies involving bone (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Single and multiple 5- and 15-minute infusions of 2, 4, 8 and 16 mg zoledronic acid in 64 patients with bone metastases yielded the following pharmacokinetic data, which were found to be dose-independent.
After initiating the infusion of zoledronic acid, the plasma concentrations of zoledronic acid rapidly increased, achieving their peak at the end of the infusion period, followed by a rapid decline to <10% of peak after 4 hours and <1% of peak after 24 hours, with a subsequent prolonged period of very low concentration not exceeding 0.1% of peak prior to the second infusion of zoledronic acid on day 28.
Intravenously administered zoledronic acid is eliminated by a triphasic process:
rapid biphasic disappearance from the systemic circulation, with half-lives of t
½α 0.24 and t
½β 1.87 hours, followed by a long elimination phase with a terminal elimination half-life of t
½γ 146 hours. There was no accumulation of zoledronic acid in plasma after multiple doses of the drug given every 28 days. Zoledronic acid is not metabolised and is excreted unchanged via the kidney. Over the first 24 hours, 39 ± 16% of the administered dose is recovered in the urine, while the remainder is principally bound to bone tissue. From the bone tissue, it is released very slowly back into the systemic circulation and eliminated via the kidney. The total body clearance is 5.04 ± 2.5 L/hr, independent of dose, and unaffected by gender, age, race and body weight. Increasing the infusion time from 5 to 15 minutes caused a 30% decrease in zoledronic acid concentration at the end of the infusion, but had no effect on the area under the plasma concentration versus time curve.
The interpatient variability in pharmacokinetic parameters for zoledronic acid was high, as seen with other bisphosphonates.
No pharmacokinetic data for zoledronic acid are available in patients with hypercalcaemia or in patients with hepatic insufficiency. Zoledronic acid does not inhibit human P450 enzymes
in vitro, shows no biotransformation and in animal studies <3% of the administered dose was recovered in the faeces, suggesting no relevant role of liver function in the pharmacokinetics of zoledronic acid.
The renal clearance of zoledronic acid was correlated with creatinine clearance, renal clearance representing 75 ± 33% of the creatinine clearance, which showed a mean of 84 ± 29 mL/min (range 22 to 143 mL/min) in the 64 cancer patients studied. Population analysis showed that for a patient with creatinine clearance of 20 mL/min (severe renal impairment) or 50 mL/min (moderate impairment), the corresponding predicted clearance of zoledronic acid would be 37% or 72%, respectively, of that of a patient showing creatinine clearance of 84 mL/min. Only limited pharmacokinetic data are available in patients with severe renal insufficiency (creatinine clearance <30 mL/min).
In an
in vitro study, zoledronic acid showed low affinity for the cellular components of human blood, with a mean blood to plasma concentration ratio of 0.59 in a concentration range of 30 ng/mL to 5000 ng/mL. The plasma protein binding is low, with the unbound fraction ranging from 60% at 2 ng/mL to 77% at 2000 ng/mL of zolendronic acid.
Special populations: Paediatric patients: Limited pharmacokinetic data in children with severe osteogenesis imperfecta suggest that zoledronic acid pharmacokinetics in children 3 to 17 years are similar to those in adults at a similar mg/kg dose level. Age, body weight, gender and creatinine clearance appear to have no effect on zoledronic acid systemic exposure.
Toxicology: Preclinical Safety Data: Acute toxicity: The highest non-lethal single intravenous dose was 10 mg/kg bodyweight in mice and 0.6 mg/kg in rats.
Subchronic and chronic toxicity: Zoledronic acid was well tolerated when administered subcutaenously to rats and intravenously to dogs at doses up to 0.02 mg/kg daily for 4 weeks. Administration of 0.001 mg/kg/day subcutaneously in rats and 0.005 mg/kg intravenously once every 2-3 days in dogs for up to 52 weeks was also well tolerated.
The most frequent finding in repeat-dose studies consisted of increased primary spongiosa in the metaphyses of long bones in growing animals at nearly all doses, a finding that reflected the compound's pharmacological antiresorptive activity.
The safety margins relative to renal effects were narrow in the long-term repeat-dose parenteral animal studies but the cumulative no adverse event levels (NOAELs) in the single dose (1.6 mg/kg) and multiple dose studies of up to one month (0.06-0.6 mg/kg/day) did not indicate renal effects at doses equivalent to or exceeding the highest intended human therapeutic dose. Longer-term repeat administration at doses bracketing the highest intended human therapeutic dose of zoledronic acid produced toxicological effects in other organs, including the gastrointestinal tract, liver, spleen and lungs, and at intravenous injection sites.
Reproduction toxicity: Zoledronic acid was teratogenic in rat at subcutaneous doses ≥0.2 mg/kg. Although no teratogenicity or foetotoxicity was observed in the rabbit, maternal toxicity was found. Dystocia was observed at the lowest dose (0.01 mg/kg bodyweight) tested in the rat.
Mutagenicity and carcinogenic potential: Zoledronic acid was not mutagenic in the mutagenicity tests performed and carcinogenicity testing did not provide any evidence of carcinogenic potential.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image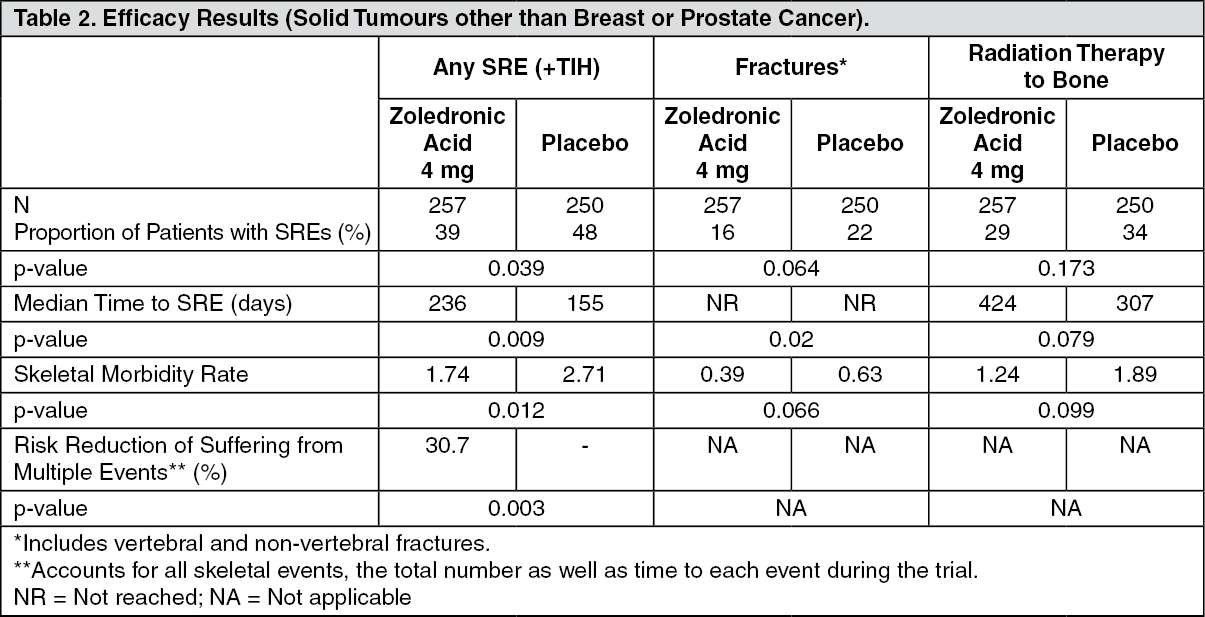 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image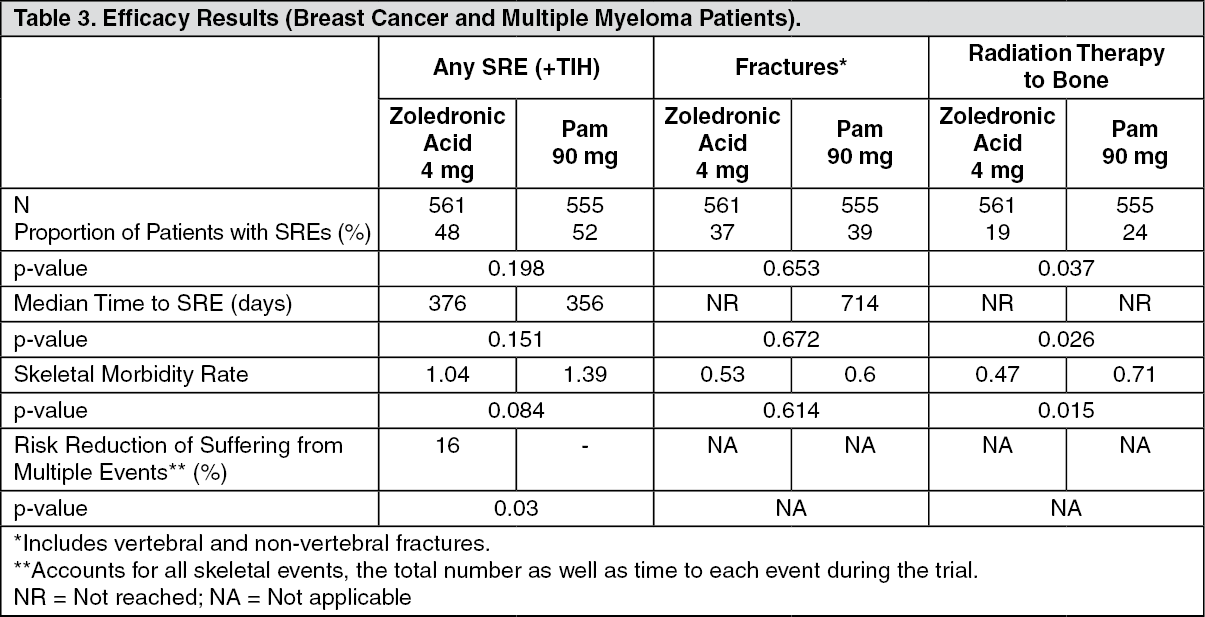 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image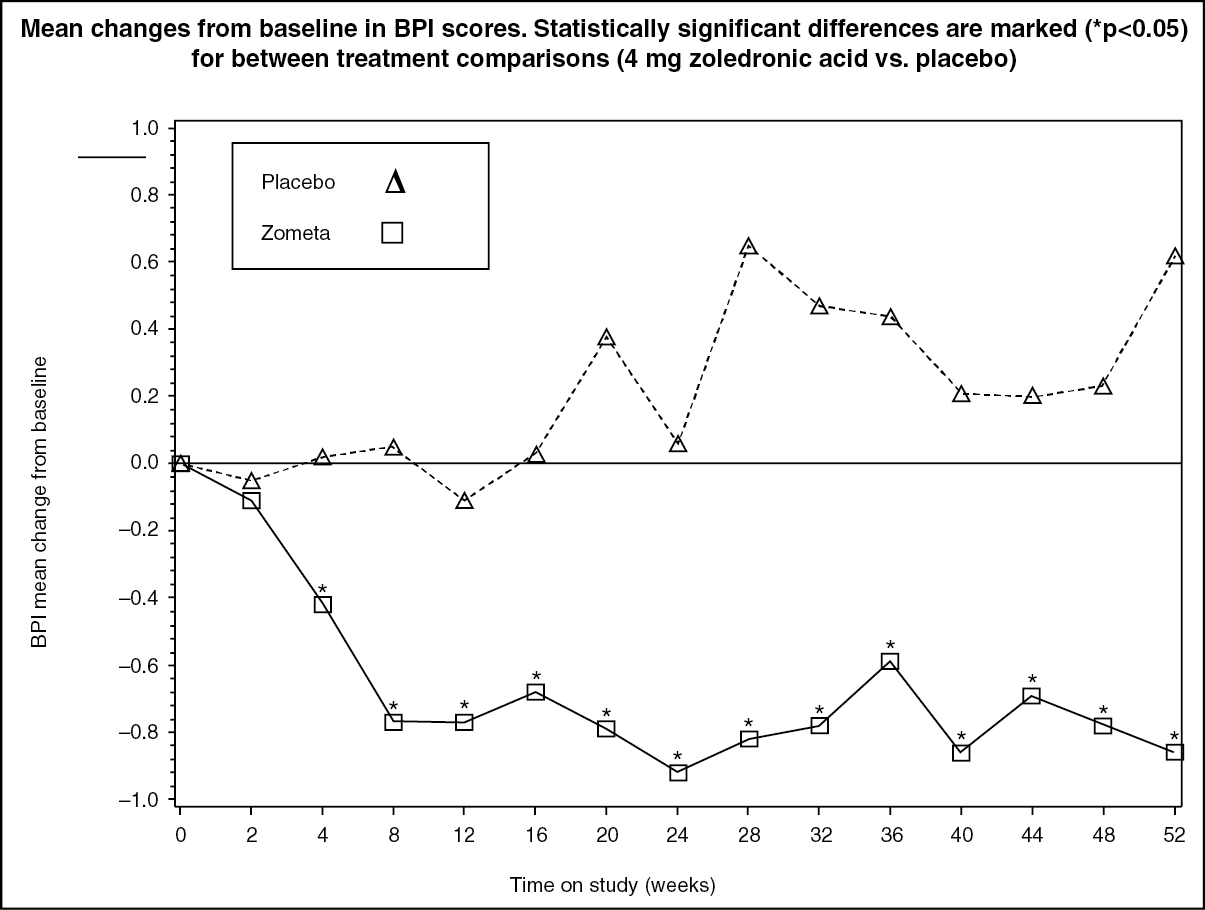 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image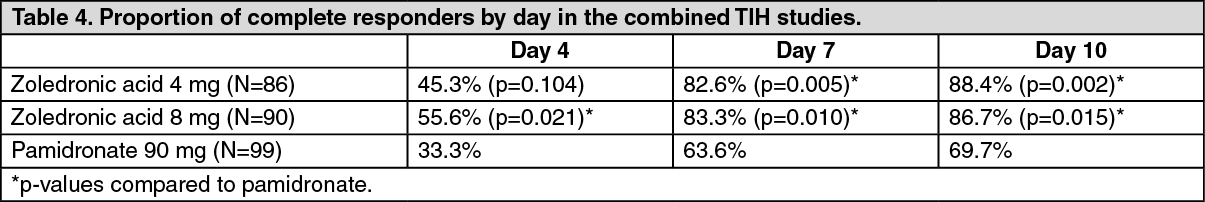 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image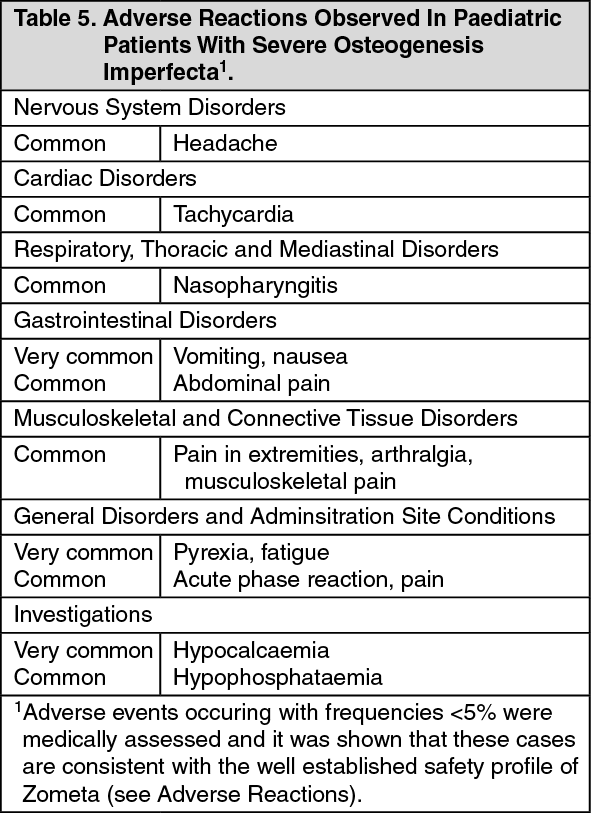 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image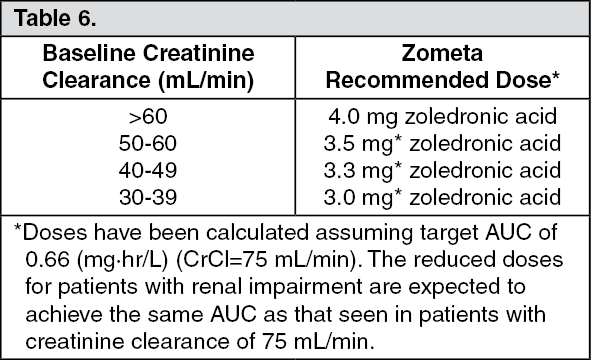 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image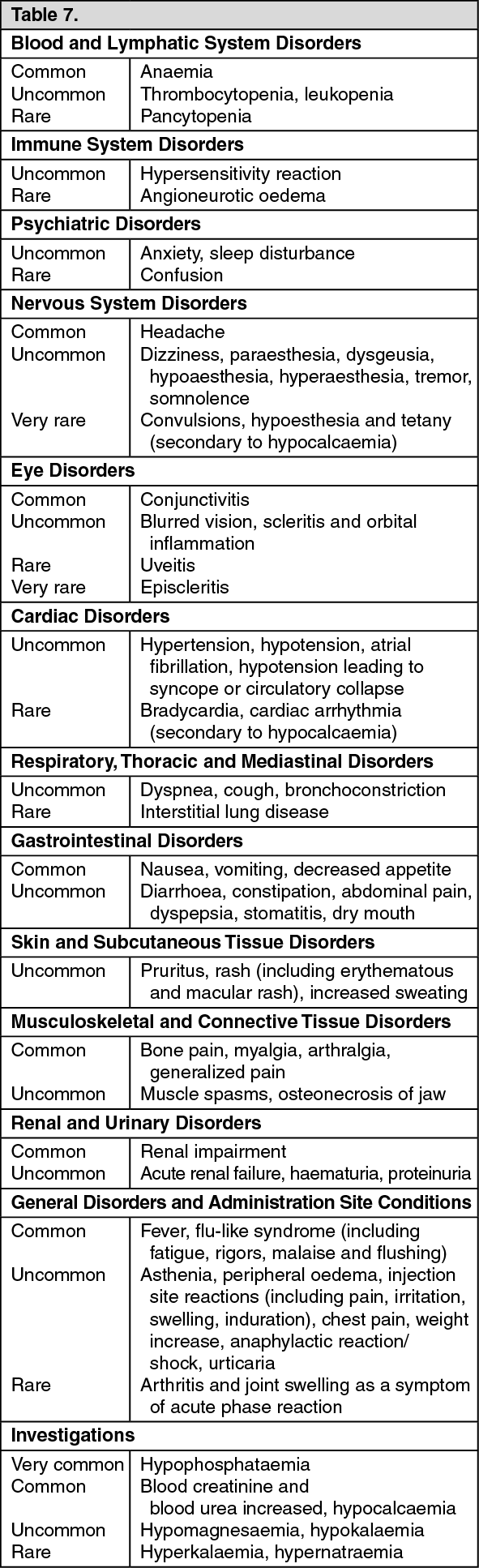 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out