


 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Mechanism of action: NOCDURNA contains desmopressin a synthetic analogue of naturally occurring anti-diuretic hormone arginine vasopressin (AVP). Desmopressin mimics vasopressin's anti-diuretic effect, binding to the V2 receptors in the renal collecting tubules of the kidneys, causing reabsorption of water into the body. This reabsorption in turn decreases night-time urine production. Due to the proposed low gender-specific doses (25 mcg for females and 50 mcg for males), and the limited duration of action of NOCDURNA, the antidiuretic activity is limited to the night-time sleep period.
Pharmacodynamic effects: In study CS29, the weight-corrected NOCDURNA dose that induced 50% maximum achievable drug effect on nocturnal urine volume differed significantly between females and males. The estimated exposure value for males was 2.7-fold (95% CI: 1.3-8.1) higher than the value for females to obtain an identical dynamic effect, corresponding to higher desmopressin sensitivity among females. The development of hyponatremia is dose dependent. Females are at higher risk of developing hyponatraemia than males. The incidences of hyponatremia rises with increasing age (see Dosage & Administration and Precautions).
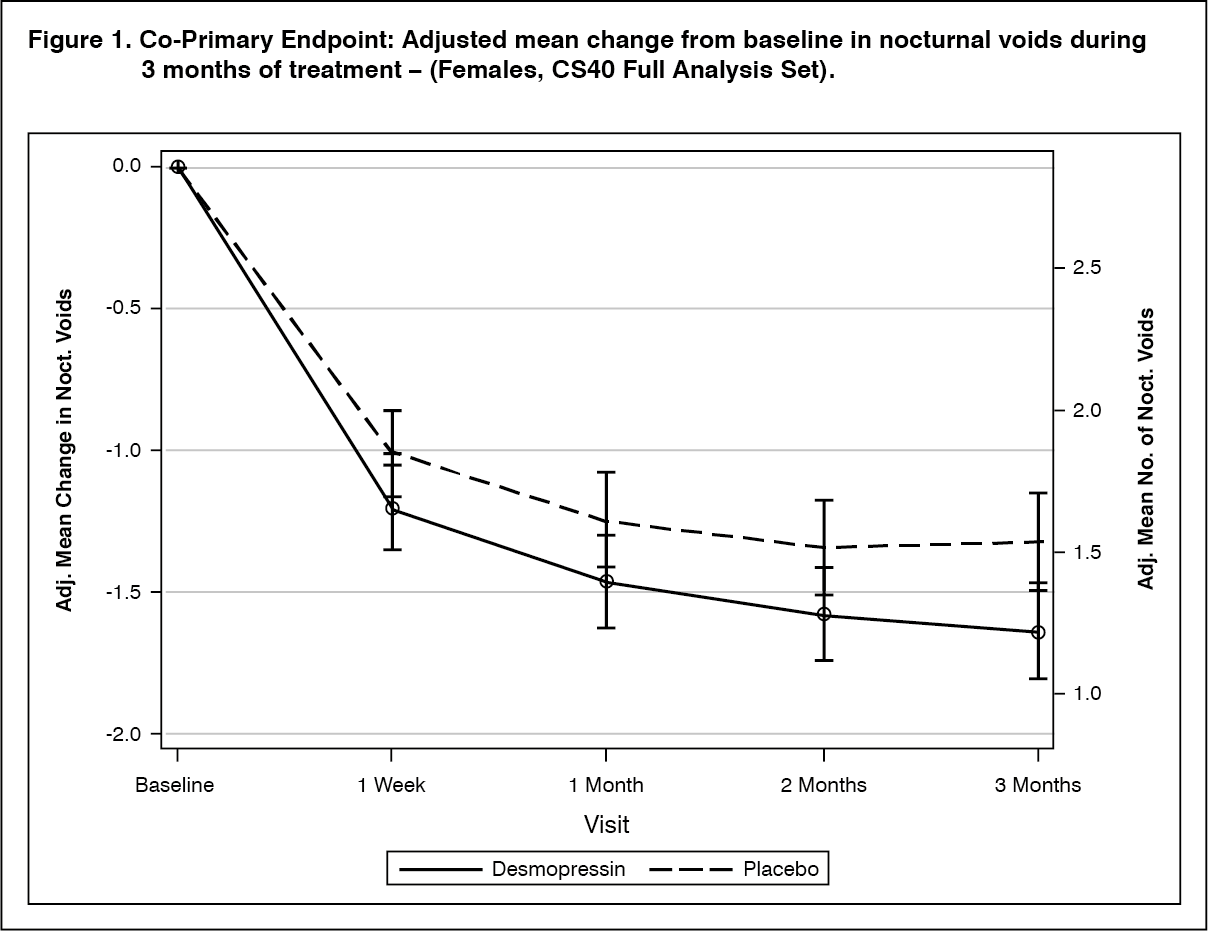
Clinical efficacy: The efficacy of NOCDURNA has been demonstrated in two randomised double blinded placebo controlled studies in respectively 268 women (study CS40, desmopressin oral lyophilisate 25 mcg versus placebo) and 395 men (study CS41, desmopressin oral lyophilisate 50 mcg and 75 mcg versus placebo) with nocturia defined as an average of ≥2 nocturnal voids per night and polyuria in 90% of women and 87% of men.
Both studies met the 2 co-primary endpoints with statistically significant differences favouring desmopressin oral lyophilisate over the 3-month period. There was a statistically significant decrease in the adjusted mean number of nocturnal voids from the baseline on desmopressin oral lyophilisate 25 mcg (-1.46) compared to placebo (-1.24) in the female study (p=0.028) (Fig. 1) and on desmopressin oral lyophilisate 50 mcg (-1.25) compared to placebo (-0.88) in the male study (p=0.0003) (Fig. 2). The proportion of subjects with >33% decrease in the mean number of nocturnal voids (responders) was significantly increased, nearly doubled. The odds ratio for >33% decrease of desmopressin oral lyophilisate 25 mcg compared to placebo was 1.85 (p=0.006) in the female study and the odds ratio for >33% decrease of desmopressin oral lyophilisate 50 mcg compared to placebo was 1.98 (p=0.0009) in the male study.
For secondary endpoints, there was an increase from baseline to 3 months in the first undisturbed sleep period (FUSP)/time to first void with a treatment contrast of 49 minutes in the female study and 39 minutes in the male study. There was a statistically significant improvement in quality of life for desmopressin oral lyophilisate 25 mcg (N-QoL total score 27.24) compared to placebo (21.90) (p=0.0226) in female and an improvement for desmopressin oral lyophilisate 50 mcg (N-QoL total score 18.37) compared to placebo (13.88) (p=0.0385) in male. There was a strong association (p<0.0001) in the both studies between treatment response (reduction in number of nocturnal voids and increase in FUSP) and improvements in patients' quality of life. (See Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image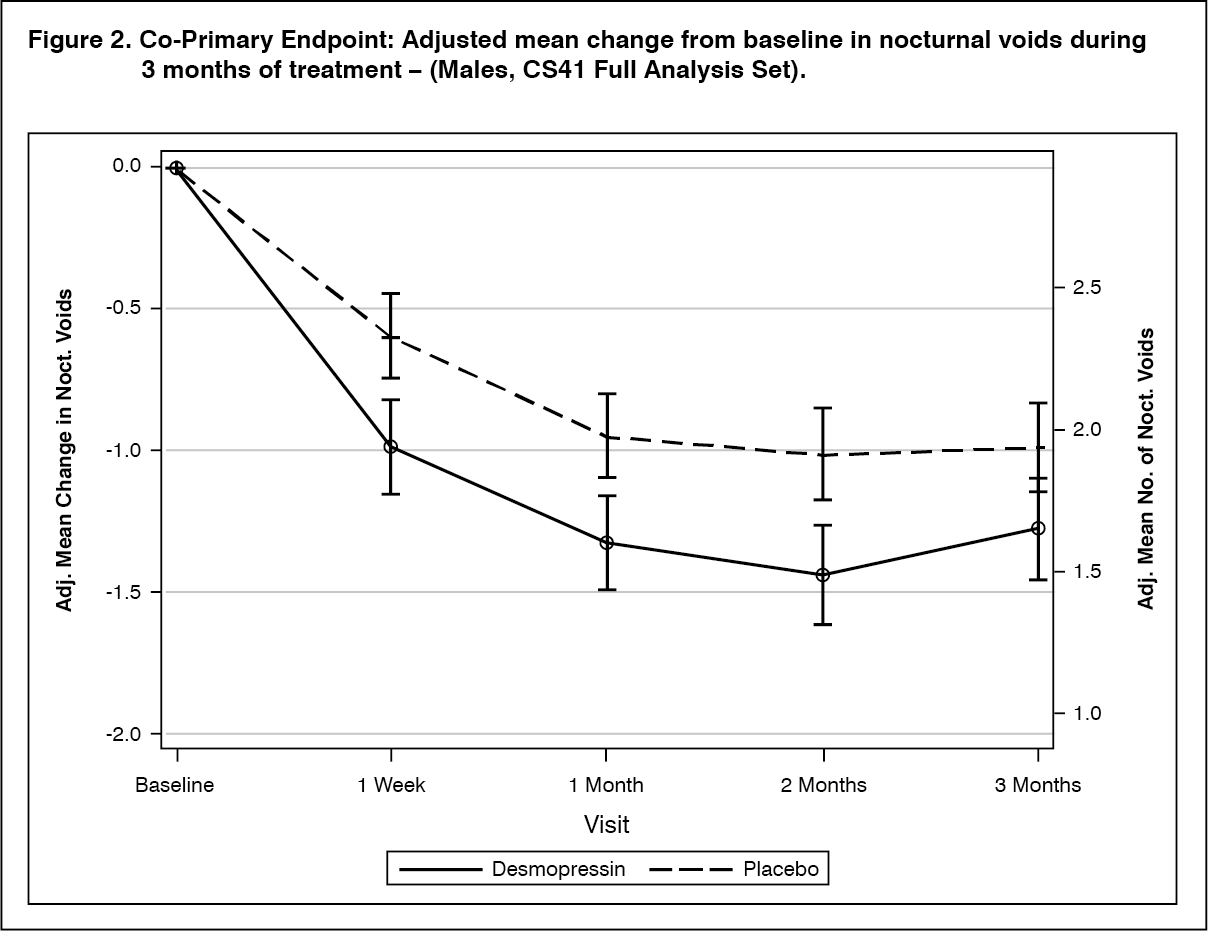 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageGender differences in clinical safety and efficacy: Clinical study [FE992026 CS029] analysed the dose-response to NOCDURNA in females and males at doses ranging from 10 to 100 mcg: In females, there was no further gain in pharmacodynamic effect above the dose of 25 mcg, indicating that the dose response plateau was reached at 25 mcg in females. In males, reduction in urine volume was greater at 50 mcg, but not substantially higher at 100 mcg. Increasing doses to 50 mcg dose level in females did not yield further efficacy, but was associated with a 5-fold increase in the risk of hyponatraemia compared with males in the age group above 50 years (p=0.015).
Pharmacokinetics: Absorption: The overall mean absolute bioavailability of desmopressin administered sublingually from earlier dose-ranging studies of doses of 200, 400 and 800 mcg is 0.25%, with a 95% confidence interval of 0.21-0.31%. Desmopressin exhibits a moderate-to-high variability in bioavailability, both within and between subjects. Desmopressin shows dose linearity regarding AUC and Cmax in the range of 60 to 240 mcg. However, the bioavailability of doses below 60 has not been evaluated.
Distribution: The distribution of desmopressin is best described by a two-compartment distribution model with a volume of distribution during the elimination phase of 0.3-0.5 L/kg.
Biotransformation: The in-vivo metabolism of desmopressin has not been studied. In vitro human liver microsome metabolism studies of desmopressin have shown that no significant amount is metabolized in the liver by the cytochrome P450 system. Thus human liver metabolism in vivo by the cytochrome P450 system is unlikely to occur. The effect of desmopressin on the PK of other drugs is likely to be minimal due to its lack of inhibition of the cytochrome P450 drug metabolizing system.
Elimination: The total clearance of desmopressin has been calculated to 7.6 L/hr. The terminal half-life of desmopressin is estimated to 2.8 hours. In healthy subjects the fraction excreted unchanged was 52% (44%-60%).
Linearity/non-linearity: There are no indications of non-linearities in any of the pharmacokinetic parameters of desmopressin.
Characteristics in specific groups of patients: Renal impairment: Depending on the degree of renal impairment the AUC and half-life increased with the severity of the renal impairment. Desmopressin is contraindicated in patients with moderate and severe renal impairment (creatinine clearance below 50 ml/min). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHepatic impairment: No studies have been performed in this population.
It is unlikely that desmopressin will interact with drugs affecting hepatic metabolism, since desmopressin has been shown not to undergo significant liver metabolism in in vitro studies with human microsomes.
Toxicology: Preclinical Safety Data: Non-clinical data revealed no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and toxicity to reproduction.
Carcinogenicity studies have not been performed with desmopressin, because it is closely related to the naturally-occurring peptide hormone.