


 Sign Out
Sign Out

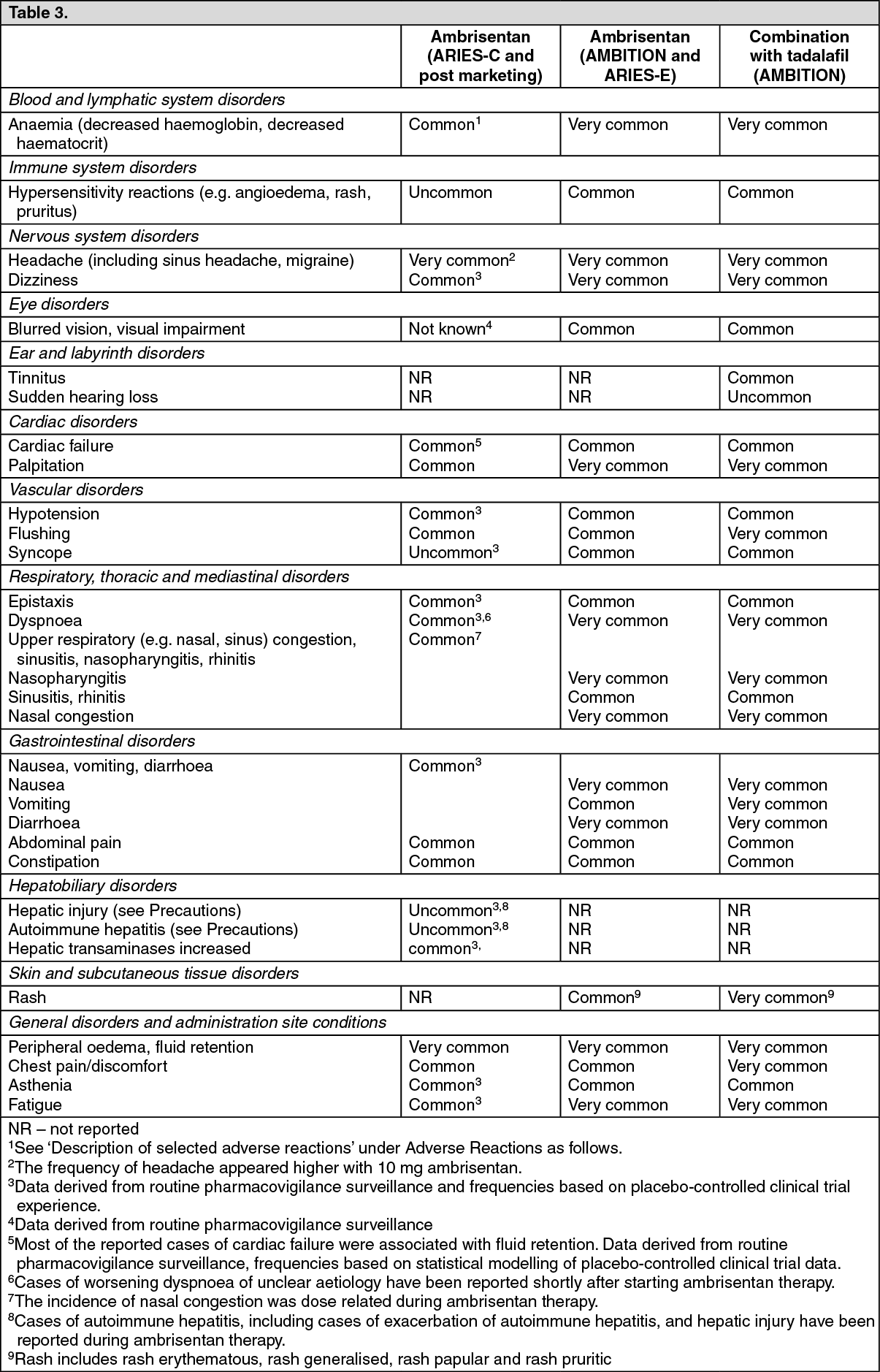
Peripheral oedema, fluid retention and headache (including sinus headache, migraine) were the most common adverse reactions observed with ambrisentan. The higher dose (10 mg) was associated with a higher incidence of these adverse reactions, and peripheral oedema tended to be more severe in patients ≥65 years in short-term clinical studies (see Precautions).
Tabulated list of adverse reactions: Frequencies are defined as: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000) and not known (cannot be estimated from available data). For dose-related adverse reactions, the frequency category reflects the higher dose of ambrisentan. Frequency categories do not account for other factors including varying study duration, pre-existing conditions and baseline patient characteristics. Adverse reaction frequency categories assigned based on clinical trial experience may not reflect the frequency of adverse events occurring during normal clinical practice. Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Decreased haemoglobin: In the post-marketing period, cases of anaemia requiring blood cell transfusion have been reported (see Precautions). The frequency of decreased haemoglobin (anaemia) was higher with 10 mg ambrisentan. Across the 12 week placebo-controlled Phase 3 clinical studies, mean haemoglobin concentrations decreased for patients in the ambrisentan groups and were detected as early as week 4 (decrease by 0.83 g/dL); mean changes from baseline appeared to stabilise over the subsequent 8 weeks. A total of 17 patients (6.5%) in the ambrisentan treatment groups had decreases in haemoglobin of ≥15% from baseline and which fell below the lower limit of normal.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
View ADR Monitoring Form