


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of action: Abrocitinib (Cibinqo) is a Janus kinase (JAK) 1 inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within signaling pathways, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Inhibition of JAK1 modulates the signaling pathway, by preventing the phosphorylation and activation of STATs.
In biochemical assay, Abrocitinib has selectivity for JAK1 over the other 3 JAK isoforms JAK2 (28-fold), JAK3 (>340-fold) and tyrosine kinase 2 (TYK 2, 43-fold). In cellular settings, it preferentially inhibits cytokine-induced STAT phosphorylation by signaling pairs involving JAK1, and spares signaling by JAK2/JAK2 or JAK2/TYK2 pairs. The relevance of selective enzymatic inhibition of specific JAK enzymes to clinical effect is not currently known.
Pharmacodynamic effects: Treatment with Abrocitinib (Cibinqo) was associated with dose-dependent reduction in serum markers of inflammation, including high sensitivity C-reactive protein (hsCRP), interleukin-31 (IL-31) and thymus and activation-regulated chemokine (TARC). These changes returned to near baseline within 4 weeks of drug discontinuation.
Mean absolute lymphocyte count increased by 2 weeks after starting treatment with Abrocitinib and returned to baseline by Month 9 of treatment. Most patients maintained an ALC within the reference range. Treatment with Abrocitinib was associated with a dose-related increase in B cell counts and a dose-related decrease in NK cell counts. The clinical significance of these changes in B cell and NK cell counts is unknown.
Clinical efficacy and safety: The efficacy and safety of Abrocitinib (Cibinqo) as monotherapy and in combination with background medicated topical therapies over 12-16 weeks were evaluated in 1616 patients in 3 pivotal randomized, double-blind, placebo controlled studies (MONO-1, MONO 2, and COMPARE). In addition, the efficacy and safety of Abrocitinib (Cibinqo) in monotherapy over 52 weeks (with the option of rescue treatment in flaring patients) was evaluated in 1233 patients in a Phase 3 induction, randomized withdrawal, double-blind, placebo-controlled study (REGIMEN). The patients in these 4 studies were 12 years of age and older with moderate-to-severe atopic dermatitis as defined by Investigator's Global Assessment (IGA) score ≥3, Eczema Area and Severity Index (EASI) score ≥16, body surface area (BSA) involvement ≥10%, and Peak Pruritus Numerical Rating Scale (PP-NRS) ≥4 at the baseline visit prior to randomization. Patients who had a prior inadequate response or for whom topical treatments were medically unadvisable, or who had received systemic therapies were eligible for inclusion.
All patients who completed the parent studies were eligible to enroll into the long-term extension study EXTEND.
Clinical response: Treatment with Abrocitinib (Cibinqo) 100 mg or 200 mg once daily as monotherapy or in combination with background medicated topical therapy resulted in improvement in objective signs of atopic dermatitis and patient-reported pruritus.
Baseline characteristics: In the placebo-controlled studies (MONO-1, MONO-2, COMPARE) and the open label induction, randomized withdrawal study (REGIMEN) across all treatment groups 41.4% to 51.1% were female, 59.3% to 77.8% were Caucasian, 15.0% to 33.0% were Asian and 4.1% to 8.3% were Black, and the mean age was 32.1 to 37.7 years. In these studies, 32.2% to 40.8% had a baseline IGA of 4 (severe atopic dermatitis), and 41.4% to 59.5% of patients had received prior systemic treatment for atopic dermatitis. The baseline mean EASI score ranged from 28.5 to 30.9, the baseline PP-NRS ranged from 7.0 to 7.3 and the baseline Dermatology Life Quality Index (DLQI) ranged from 14.4 to 16.0.
Monotherapy studies: In both pivotal monotherapy studies (MONO-1, MONO-2), the proportion of patients who achieved IGA and/or EASI-75 response was significantly higher in patients who received Abrocitinib (Cibinqo) 100 mg or 200 mg once daily compared with placebo at Week 12 (see Table 1).
A significantly higher proportion of patients who achieved PP-NRS4 (defined as an improvement of ≥4 points in the severity of PP-NRS) with Abrocitinib (Cibinqo) 100 mg or 200 mg once daily compared with placebo was observed as soon as Week 2 and persisting through Week 12. Higher proportions of patients achieved PP-NRS4 with Abrocitinib (Cibinqo) 100 mg or 200 mg once daily compared with placebo by Day 6 and Day 3 (2 days after the first dose), respectively (see Table 1).
 Click on icon to see table/diagram/image
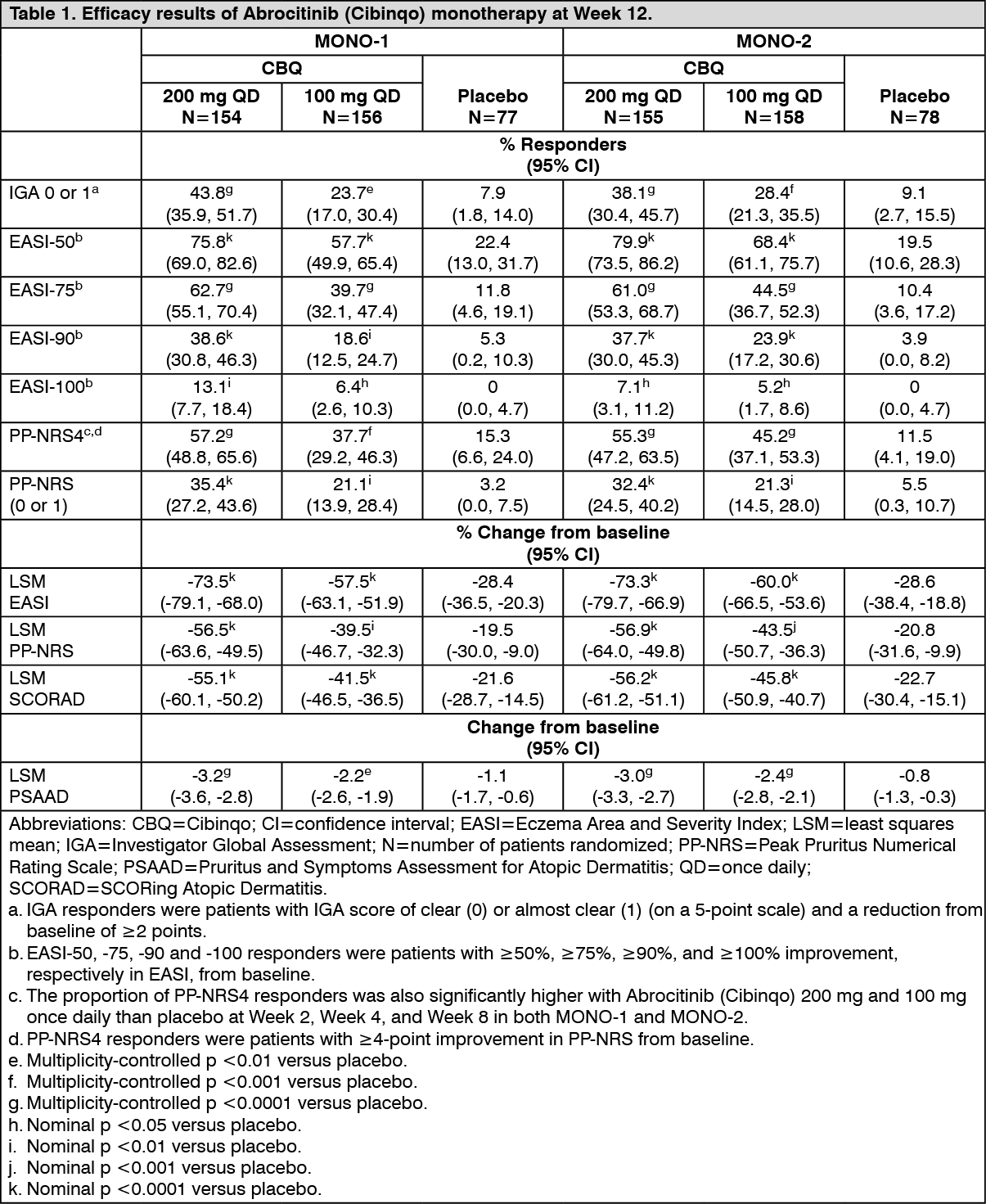
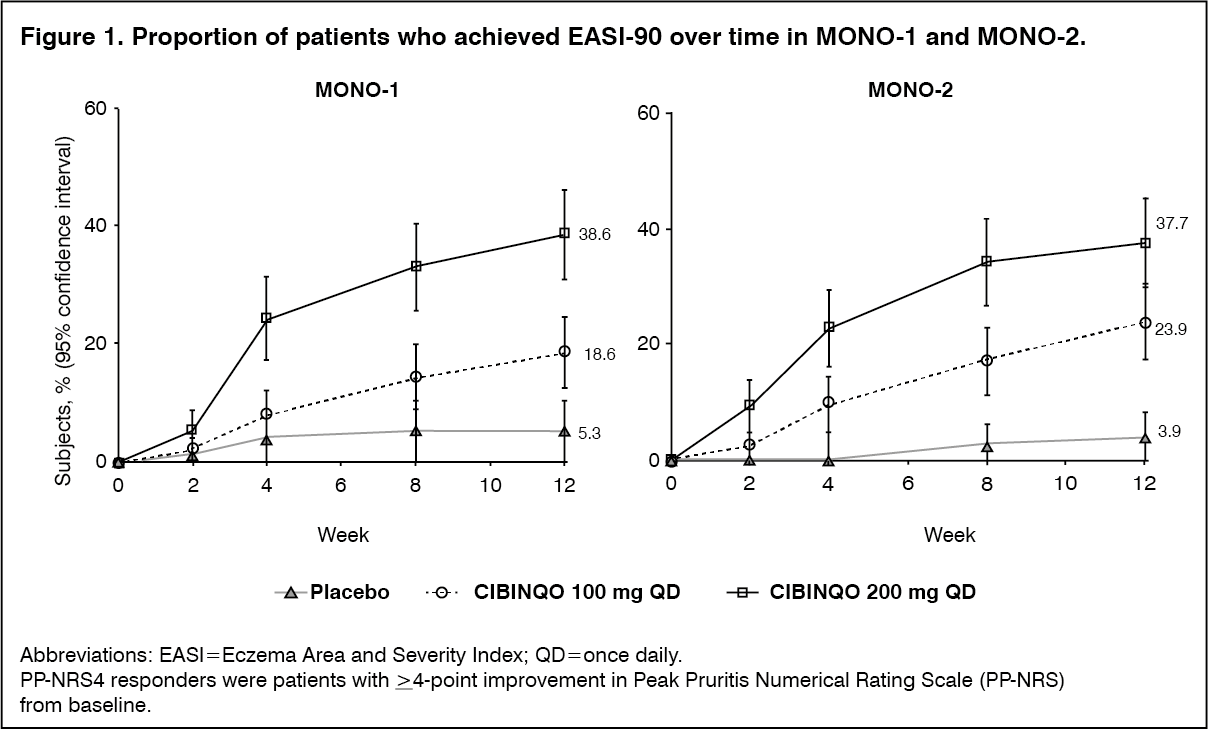
Click on icon to see table/diagram/imageThe proportion of patients who achieved EASI-90 or PP-NRS4 over time in studies MONO-1 and MONO-2 are shown in Figures 1 and 2. (See Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment effects in subgroups (e.g., weight, age, sex, race and prior systemic immunosuppressant treatment) in MONO-1 and MONO-2 were consistent with the results in the overall study population.
Combination therapy study: In the pivotal combination therapy study (COMPARE), the proportion of patients who achieved IGA or EASI-75 response was significantly higher in patients who received Abrocitinib (Cibinqo) 100 mg or 200 mg once daily compared with placebo at Week 12 (see Table 2).
The proportions of patients achieving PP-NRS4 with Abrocitinib (Cibinqo) 100 mg and 200 mg once daily were significantly higher than placebo by Day 9 and Day 4, respectively, and remained significantly higher than placebo with both Abrocitinib (Cibinqo) doses at Week 2 and Week 16.
The proportion of patients achieving PP-NRS4 with Abrocitinib (Cibinqo) 200 mg once daily was significantly higher than dupilumab as early as Day 4 and remained significantly higher than dupilumab at Week 2. The proportion of patients achieving PP-NRS4 was similar between abrocitinib (Cibinqo) 100 mg once daily and dupilumab at Week 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe proportion of patients who achieved EASI-90 or PP-NRS4 over time in COMPARE are shown in Figure 3. (See Figure 3.)
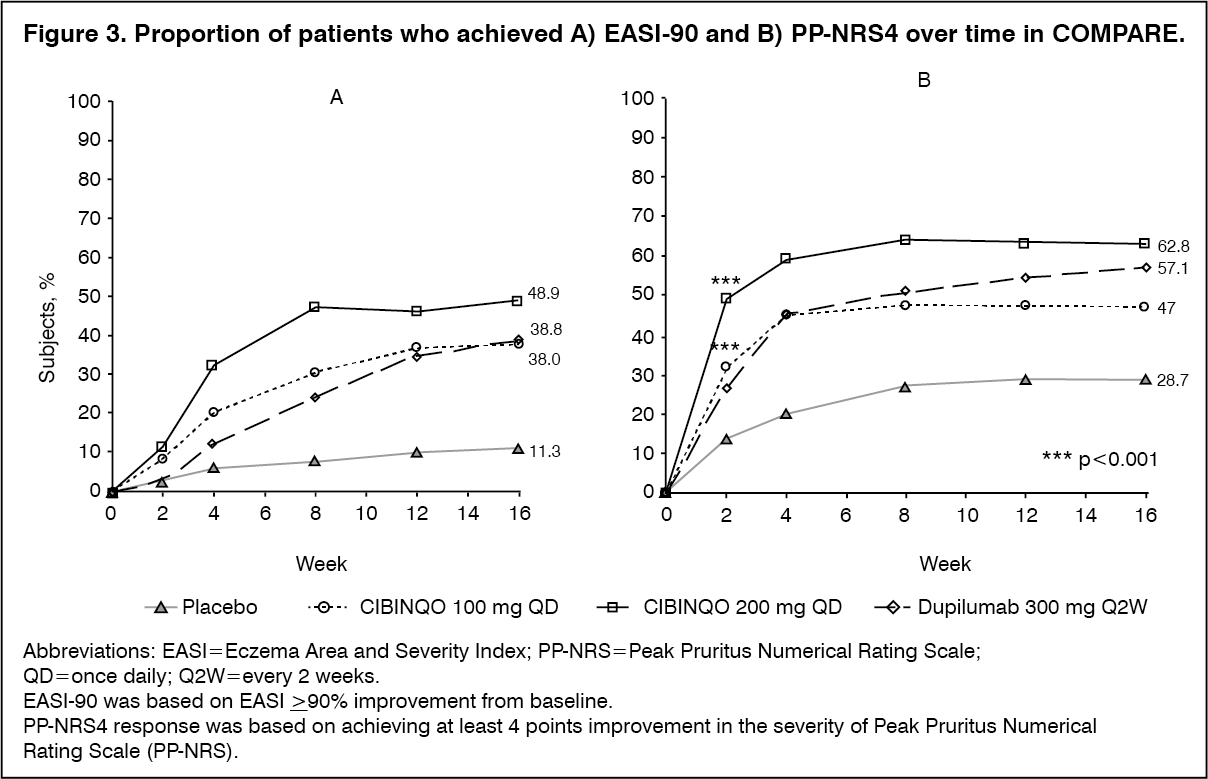 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients who received dupilumab and subsequently enrolled in EXTEND were randomized to either Abrocitinib (Cibinqo) 100 mg or 200 mg once daily upon entering EXTEND. Among responders to dupilumab in COMPARE, the majority maintained response 12 weeks after switching to Abrocitinib (Cibinqo) [77% and 86% for IGA (0 or 1) response, and 90% and 96% for EASI-75 with 100 mg once daily or 200 mg once daily, respectively]. Among non-responders to dupilumab in COMPARE, a substantial proportion of patients achieved response 12 weeks after switching to Abrocitinib (Cibinqo) [34% and 47% for IGA (0 or 1) response, and 68% and 80% for EASI-75 with 100 mg once daily or 200 mg once daily, respectively].
Treatment effects in subgroups (e.g., weight, age, sex, race, and prior systemic immunosuppressant treatment) in COMPARE were consistent with the results in the overall study population.
Late-onset efficacy: Eligible patients who completed the full treatment period of a qualifying parent study (e.g., MONO-1, MONO-2, COMPARE) were considered for enrollment in the long-term extension study EXTEND, which allows patients to extend Abrocitinib (Cibinqo) treatment for at least 92 weeks or until availability of commercial product in their country. In EXTEND, patients received Abrocitinib (Cibinqo) with or without background medicated topical therapy. Patients who were previously randomized to Abrocitinib (Cibinqo) 100 mg or 200 mg once daily in qualifying studies continued the same dose in EXTEND as in the parent study, and the blind was maintained. Patients not previously randomized to Abrocitinib (Cibinqo) in a qualifying parent study were randomized to either Abrocitinib (Cibinqo) 100 mg or 200 mg once daily upon entering EXTEND.
Among patients who did not achieve IGA (0 or 1) response after 12 weeks of Abrocitinib (Cibinqo) treatment and entered EXTEND, 14% and 22% of patients continuing Abrocitinib (Cibinqo) 100 mg once daily in EXTEND achieved IGA (0 or 1) response by Week 16 and Week 24 (with 4 and 12 additional weeks of treatment), respectively, and 19% and 27% of patients continuing Abrocitinib (Cibinqo) 200 mg once daily achieved IGA response by Week 16 and Week 24, respectively. Among patients who did not achieve EASI-75 after 12 weeks of Abrocitinib (Cibinqo) treatment and entered EXTEND, 32% and 45% of patients continuing Abrocitinib (Cibinqo) 100 mg once daily in EXTEND achieved EASI-75 by Week 16 and Week 24 (with 4 and 12 additional weeks of treatment), respectively, and 34% and 54% of patients continuing Abrocitinib (Cibinqo) 200 mg once daily achieved EASI-75 response by Week 16 and Week 24, respectively.
Long-term efficacy: Among patients who achieved response at Week 12 of a qualifying parent study and entered EXTEND, the majority of patients maintained their response at Week 48 of cumulative Abrocitinib (Cibinqo) treatment for both doses of Abrocitinib (Cibinqo) [53% and 57% for IGA (0 or 1) response, 69% and 71% for EASI-75, and 52% and 69% for PP-NRS4 with 100 mg once daily and 200 mg once daily, respectively].
Health related outcomes: Treatment with either dose of Abrocitinib (Cibinqo) as monotherapy resulted in significantly improved patient-reported outcomes at 12 weeks compared with placebo (see Table 3). A significantly larger proportion of the Abrocitinib (Cibinqo) groups had clinically meaningful reductions in Dermatology Life Quality Index (DLQI) total scores (defined as a 4-point improvement) from baseline to Week 12 compared with placebo. Abrocitinib (Cibinqo) groups also had a significantly larger proportion of patients who reported "no effect" of their disease on their quality of life (as measured by a DLQI score of 0 or 1).
Both groups significantly improved patient-reported atopic dermatitis symptoms and sleep disruption as measured by the Patient Oriented Eczema Measure (POEM), Night Time Itch Scale (NTIS), and SCORing Atopic Dermatitis (SCORAD) sleep loss subscale. In addition, anxiety and depression symptoms as measured by the Hospital Anxiety and Depression Scale (HADS) total score were significantly reduced in the Abrocitinib (Cibinqo) groups compared with placebo at 12 weeks. (See Table 3.)
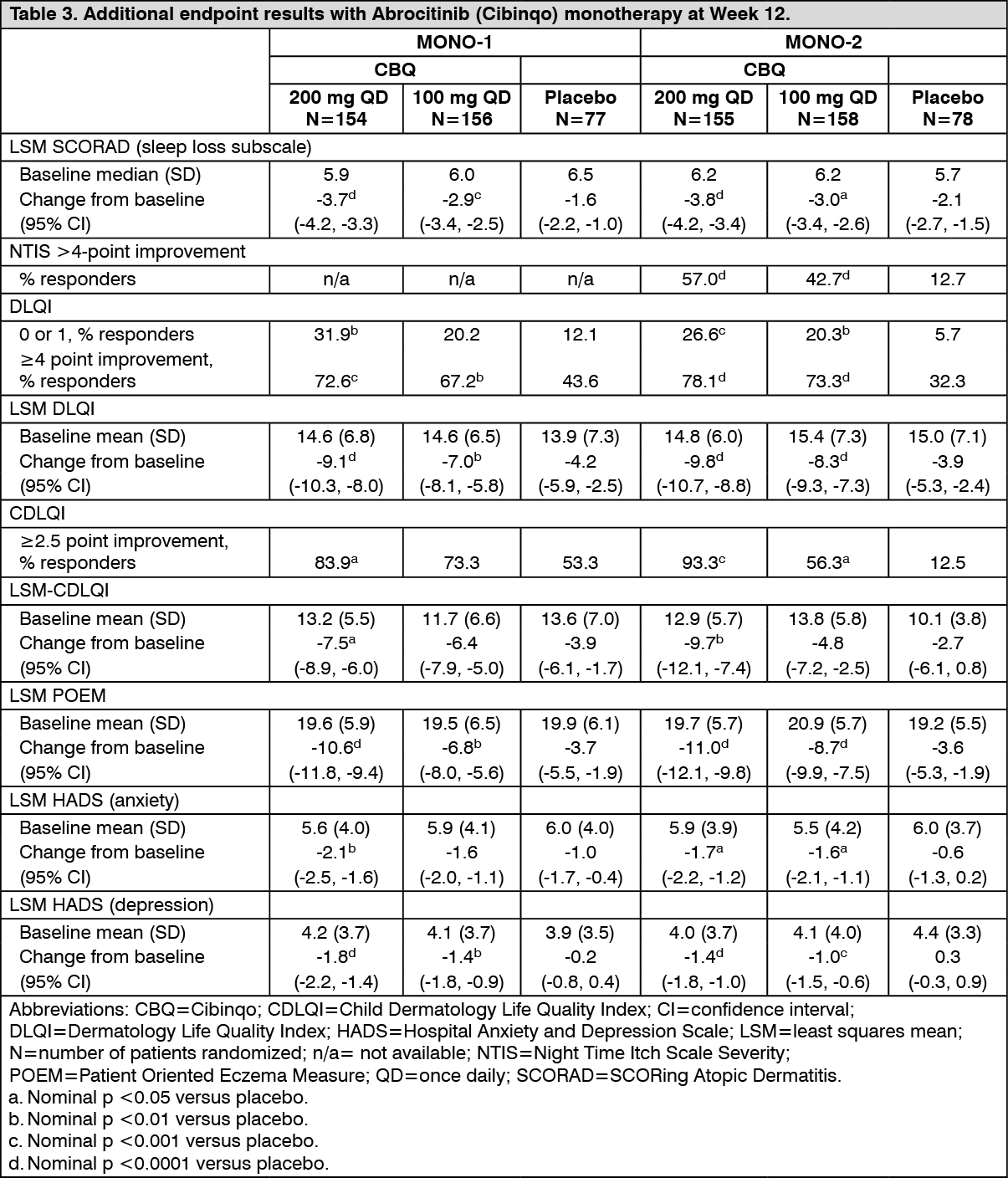 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn COMPARE, a significantly larger proportion of the Abrocitinib (Cibinqo) groups had clinically meaningful reductions in DLQI total scores (defined as a 4 point improvement) from baseline to Week 12 compared with placebo (see Table 4). Abrocitinib (Cibinqo) groups also had a significantly larger proportion of patients who reported "no effect" of their disease on their quality of life (as measured by a DLQI score of 0 or 1).
Both groups significantly improved patient-reported atopic dermatitis symptoms and sleep disruption as measured by the POEM and SCORAD sleep loss subscale, respectively. In addition, anxiety and depression symptoms as measured by the HADS total score were significantly reduced in the Abrocitinib (Cibinqo) groups compared with placebo at 12 weeks. (See Table 4.)
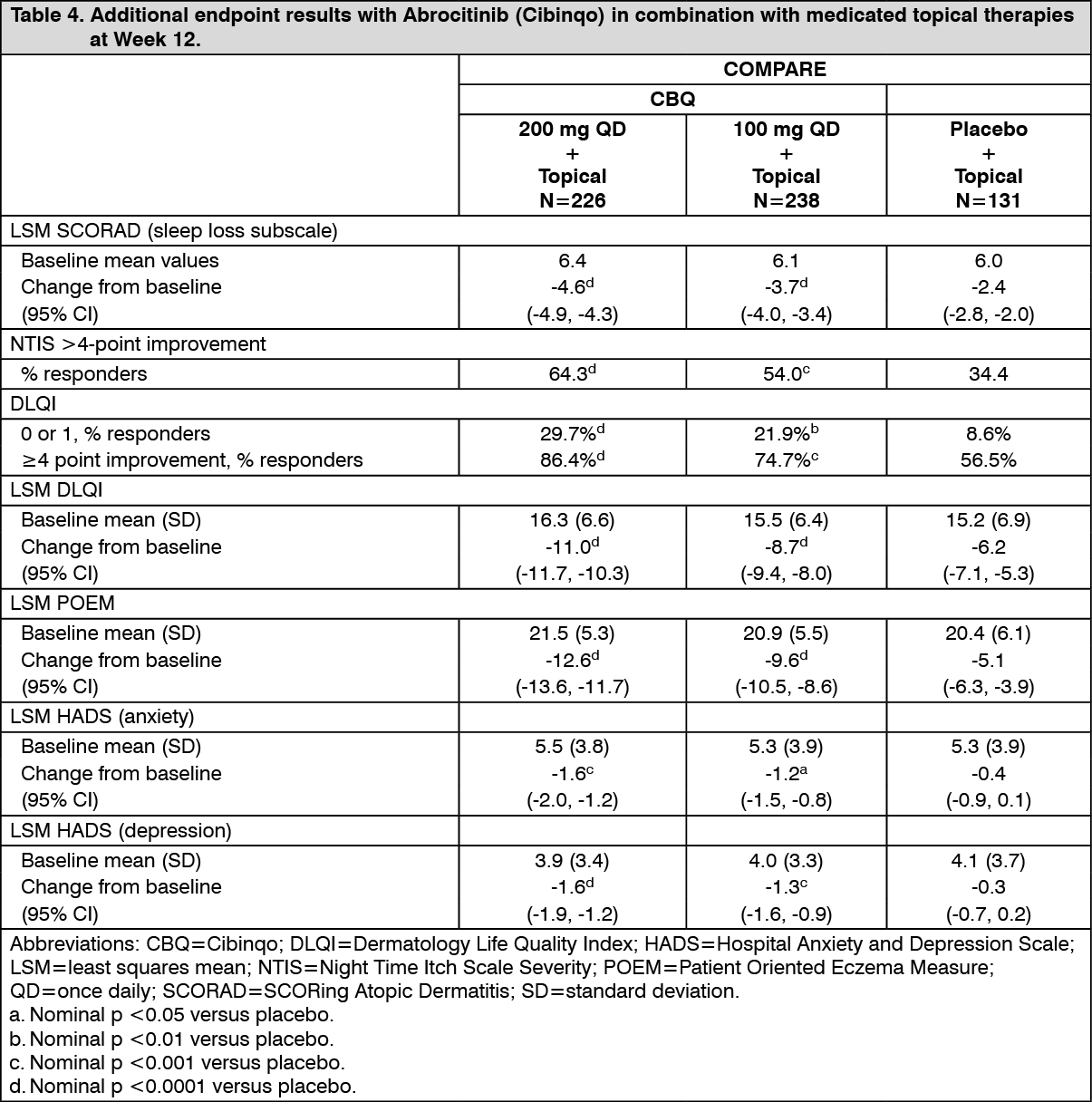 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOpen label induction, randomized withdrawal study (REGIMEN): A total of 1,233 patients received open label Abrocitinib (Cibinqo). Seven-hundred ninety-eight (798) induction responders were randomized to 200 mg or 100 mg of medicinal product or placebo.
Continuous treatment (200 mg continuous) and induction-maintenance treatment (200 mg for 12 weeks followed by 100 mg) prevented flare with 81.1% and 57.4% probability, respectively, versus 19.1% among patients who withdrew treatment (randomized to placebo) after 12 weeks of induction. Three-hundred fifty-one (351) patients including 16.2% of 200 mg, 39.2% of 100 mg and 76.4% of placebo patients received rescue medication of 200 mg Abrocitinib (Cibinqo) in combination with topical therapy. (See Table 5 and Figure 4.)
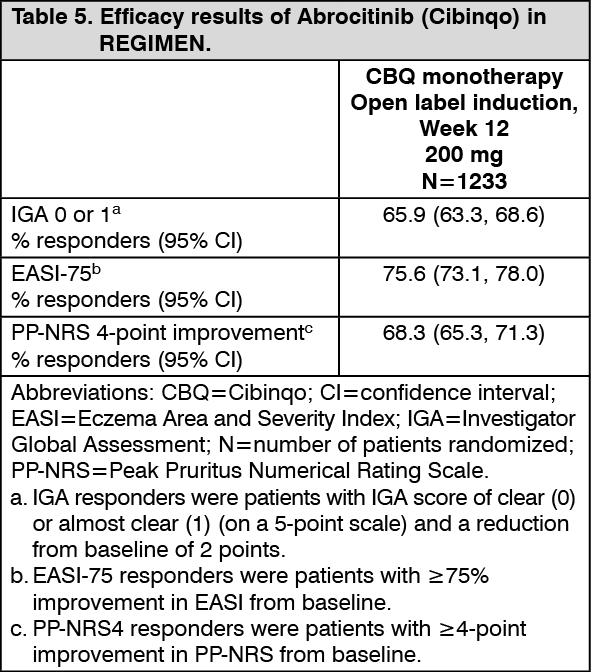 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image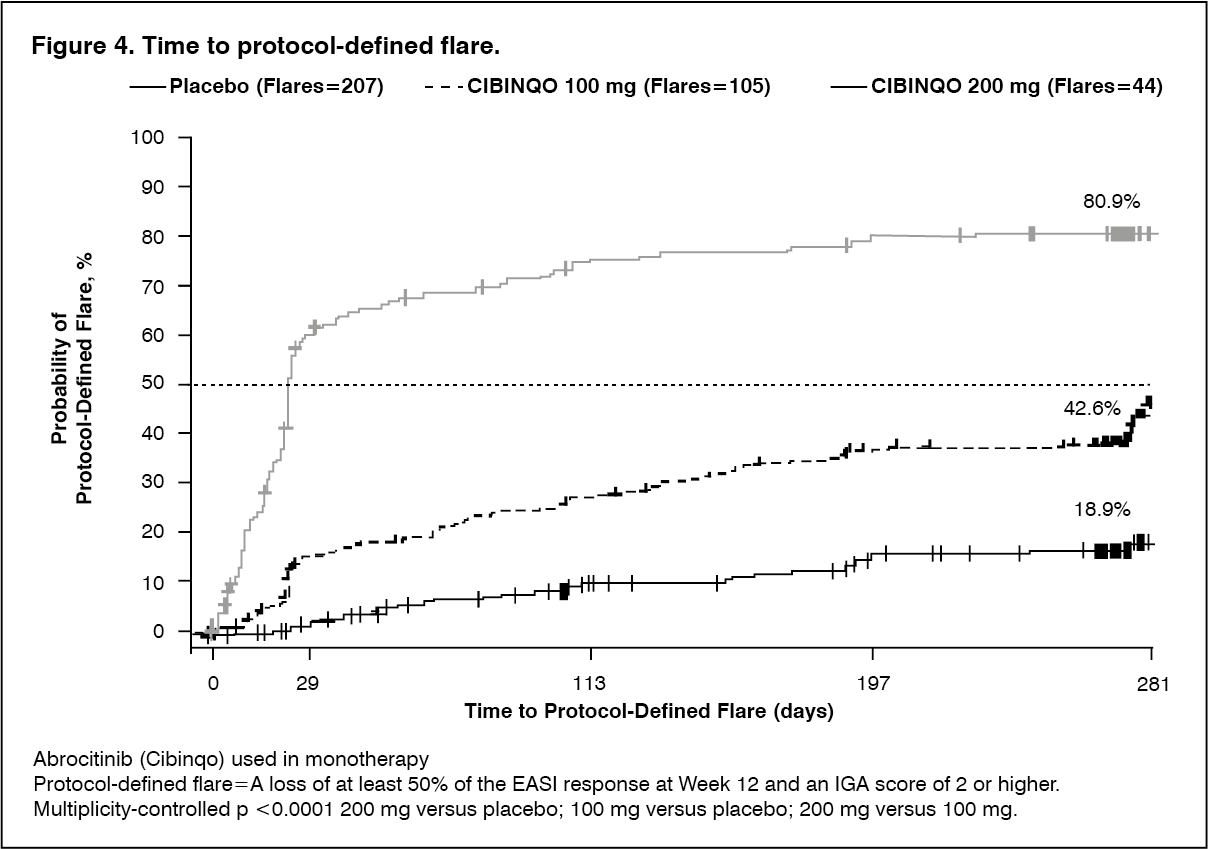 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA multivariate analysis was performed to identify predictors of successfully decreasing the dose from 200 mg to 100 mg and remaining flare-free for at least 12 weeks after the dose decrease. In that analysis, patients who had not received prior systemic agents (OR 1.8, 95% CI: 1.2, 2.6) and patients who had ≤50% BSA involvement before starting Abrocitinib (OR 1.8, 95% CI: 1.2, 2.6) were almost twice as likely to remain protocol-defined flare-free than those who had received prior systemic agents and who had ˃50% BSA involvement.
Pediatric population: The efficacy and safety of Abrocitinib (Cibinqo) as monotherapy was evaluated in 2 Phase 3 randomized, double-blind, placebo-controlled studies (MONO-1, MONO-2) which included 124 patients who were 12 to less than 18 years of age. The efficacy and safety were also evaluated in open label induction, randomized withdrawal study (REGIMEN) which included 246 patients who were 12 to less than 18 years of age. In these studies, the results in the adolescent subgroup were consistent with the results in the overall study population.
The efficacy and safety of Abrocitinib (Cibinqo) in combination with background medicated topical therapy was evaluated in the Phase 3 randomized, double-blind, placebo-controlled study TEEN. The study included 285 patients who were 12 to less than 18 years of age with moderate-to-severe atopic dermatitis as defined by IGA score ≥3, EASI score ≥16, BSA involvement ≥10%, and PP-NRS ≥ 4 at the baseline visit prior to randomization. Patients who had a prior inadequate response or who had received systemic therapy, were eligible for inclusion.
Baseline characteristics: In TEEN, across all treatment groups 49.1% were female, 56.1% were Caucasian, 33.0% were Asian and 6.0% were Black patients. The median age was 15 years and the proportion of patients with severe atopic dermatitis (IGA of 4) was 38.6%. (See Table 6.)
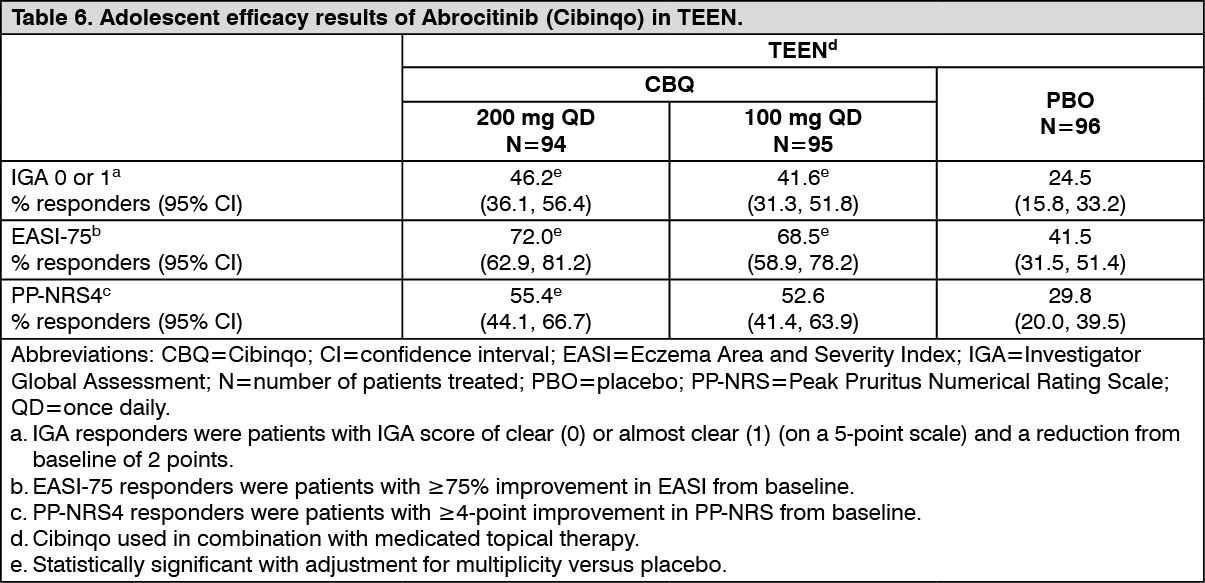 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption: Effect of Food: Abrocitinib is well-absorbed with over 91% extent of oral absorption and absolute oral bioavailability of approximately 60%. The oral absorption of Abrocitinib is rapid and peak plasma concentrations and reached within 1 hour. Steady-state plasma concentrations of Abrocitinib are achieved within 48 hours after once daily administration. Both Cmax and AUC of Abrocitinib increased dose proportionally up to 400 mg. Coadministration of Abrocitinib (Cibinqo) with a high-fat meal had no clinically relevant effect on Abrocitinib exposures (AUC and Cmax increased by approximately 26% and 29%, respectively, and Tmax was prolonged by 2 hours). In clinical studies, Abrocitinib (Cibinqo) was administered without regard to food (see Dosage & Administration).
Distribution: After intravenous administration, the volume of distribution of Abrocitinib (Cibinqo) is about 100 L. Approximately 64%, 37% and 29% of circulating abrocitinib and its active metabolites M1 and M2, respectively, are bound to plasma proteins. Abrocitinib and its active metabolites distribute equally between red blood cells and plasma.
Metabolism: The metabolism of Abrocitinib is mediated by multiple CYP enzymes, CYP2C19 (~53%), CYP2C9 (~30%), CYP3A4 (~11%) and CYP2B6 (~6%). In a human radiolabeled study, Abrocitinib was the most prevalent circulating species, with 3 polar mono-hydroxylated metabolites identified as M1 (3-hydroxypropyl), M2 (2-hydroxypropyl), and M4 (pyrrolidinone pyrimidine). Of the 3 metabolites in circulation, M1 and M2 have similar JAK inhibitory profiles as Abrocitinib, while M4 was pharmacologically inactive. The pharmacologic activity of Abrocitinib (Cibinqo) is attributable to the unbound exposures of parent molecule (~60%) as well as M1 (~10%) and M2 (~30%) in systemic circulation. The sum of unbound exposures of Abrocitinib, M1 and M2, each expressed in molar units and adjusted for relative potencies, is referred to as the Abrocitinib active moiety.
In vitro, Abrocitinib (Cibinqo) or its metabolites were not significant inhibitors or inducers of CYP enzymes (CYP2C8, CYP2C9, and CYP2D6) or of uridine diphosphate glucuronyltransferases (UGTs) (UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7). Abrocitinib or its metabolites at clinically meaningful concentrations are not inhibitors of organic anion transporter (OAT)3, organic cation transporter (OCT)1, multidrug and toxin compound extrusion protein (MATE)1/2K and breast cancer resistance protein (BCRP), organic anion transporting polypeptide (OATP) 1B1/1B3, bile salt export pump (BSEP), OAT1 or OCT2.
Elimination: The elimination half-life of Abrocitinib is about 5 hours. Abrocitinib is eliminated primarily by metabolic clearance mechanisms, with less than 1% of the dose excreted in urine as unchanged drug. The metabolites of Abrocitinib, M1, M2 and M4 are excreted predominantly in urine, and are substrates of OAT3 transporter.
Special populations: Body Weight, Gender, Genotype, Race, and Age: Body weight, gender, CYP2C19/2C9 genotype, race, and age did not have a clinically meaningful effect on Abrocitinib exposure (see Dosage & Administration).
Adolescents (12 to less than 18 years of age): Based on population pharmacokinetic analysis, there was no clinically significant difference in mean Abrocitinib steady-state exposures in adolescent patients compared to adults at their typical body weights.
Pediatric (under 12 years of age): The pharmacokinetics of Abrocitinib in pediatric patients under 12 years of age have not yet been established (see Dosage & Administration).
Renal impairment: In a renal impairment study, patients with severe (eGFR <30 mL/min) and moderate (eGFR 30 to <60 mL/min) renal impairment had approximately 191% and 110% increase in active moiety AUCinf, respectively, compared to patients with normal renal function (eGFR ≥90 mL/min; see Dosage & Administration). Pharmacokinetics of Abrocitinib have not been determined in patients with mild renal impairment, however, based on the results observed in other groups, an increase of up to 70% in active moiety exposure is expected in patients with mild renal impairment (eGFR 60 to <90 mL/min). The increase of up to 70% is not clinically meaningful as the efficacy and safety of Abrocitinib in atopic dermatitis patients with mild renal impairment (n=756) was comparable to the overall population in Phase 2 and 3 clinical studies. Based on these results, a clinically significant increase in Abrocitinib active moiety is not expected in patients with mild renal impairment (creatinine clearance 60 to <90 mL/min). The eGFR in individual patients was estimated using Modification of Diet in Renal Disease (MDRD) formula.
Abrocitinib (Cibinqo) has not been studied in patients with ESRD on renal replacement therapy (see Dosage & Administration). In Phase 3 clinical studies, Abrocitinib (Cibinqo) was not evaluated in patients with atopic dermatitis with baseline creatinine clearance values less than 40 mL/min.
Hepatic impairment: Patients with mild (Child Pugh A) and moderate (Child Pugh B) hepatic impairment had approximately 4% decrease and 15% increase in active moiety AUCinf, respectively, compared to patients with normal hepatic function. These changes are not clinically significant, and no dose adjustment is required in patients with mild or moderate hepatic impairment (see Dosage & Administration). In clinical studies, Abrocitinib (Cibinqo) was not evaluated in patients with severe (Child Pugh C) hepatic impairment, or in patients screened positive for active hepatitis B or hepatitis C.
Toxicology: Preclinical Safety Data: Genotoxicity: Abrocitinib (Cibinqo) is not mutagenic in the bacterial mutagenicity assay (Ames assay). Although Abrocitinib (Cibinqo) is aneugenic in the in vitro TK6 micronucleus assay, Abrocitinib (Cibinqo) is not aneugenic or clastogenic based on the results of the in vivo rat bone marrow micronucleus assay.
Carcinogenicity: No evidence of tumorigenicity was observed in the 6-month Tg.rasH2 mice administered Abrocitinib (Cibinqo) at oral doses up to 75 mg/kg/day and 60 mg/kg/day in female and male mice, respectively. In the 2-year oral carcinogenicity study, Abrocitinib (Cibinqo) resulted in a statistically higher incidence of benign thymomas in female rats at exposures greater than or equal to 2.8 times the unbound human AUC at the MRHD of 200 mg. No evidence of Abrocitinib (Cibinqo) related tumorigenicity was observed following oral Abrocitinib (Cibinqo) administration in female rats at exposures equal to 0.6 times the unbound human AUC at the MRHD of 200 mg or in male rats at exposures equal to 14 times the unbound human AUC at the MRHD of 200 mg.
Reproductive and developmental toxicity: Abrocitinib (Cibinqo) had no effects on rat male fertility or spermatogenesis at doses up to 70 mg/kg/day at exposures equal to 26 times the unbound human AUC at the MRHD of 200 mg. Abrocitinib (Cibinqo) resulted in effects on rat female fertility (lower fertility index, corpora lutea, and implantation sites) at exposures equal to 29 times the unbound human AUC at the MRHD of 200 mg and higher postimplantation loss at exposures greater than or equal to 11 times the unbound human AUC at the MRHD of 200 mg. The effects on female fertility reversed 1 month after cessation of Abrocitinib (Cibinqo) administration. No effects on female fertility were noted at exposures equal to 2 times the unbound human AUC at the MRHD of 200 mg.
No fetal malformations were observed in embryo-fetal development studies in rats or rabbits. In an embryo-fetal development study in pregnant rabbits, oral administration of Abrocitinib (Cibinqo) during gestation days 7 to 19 had no effects on embryo-fetal survival or fetal morphological development at exposures equal to 4 times the unbound human AUC at the MRHD of 200 mg. Abrocitinib (Cibinqo) resulted in increased incidence of delayed ossification of the forelimb phalanges at exposures equal to 4 times the unbound human AUC at the MRHD of 200 mg.
In an embryo-fetal development study in pregnant rats, oral administration of Abrocitinib (Cibinqo) during gestation days 6 to 17 resulted in increased embryo-fetal lethality at exposures equal to 17 times the unbound human AUC at the MRHD of 200 mg. No embryo-fetal lethality was observed in pregnant rats orally dosed with Abrocitinib (Cibinqo) during organogenesis at exposures equal to 11 times the unbound human AUC at the MRHD of 200 mg. Abrocitinib (Cibinqo) resulted in increased incidences of skeletal variations of short 13th ribs at exposures greater than or equal to 11 times the unbound human AUC at the MRHD of 200 mg and reduced ventral processes, thickened ribs, and unossified metatarsals at exposures equal to 17 times the unbound human AUC at the MRHD of 200 mg. No skeletal variations were noted in rats at exposures equal to 2.4 times the unbound human AUC at the MRHD of 200 mg.
In a pre- and postnatal development study in pregnant rats, oral administration of Abrocitinib (Cibinqo) during gestation day 6 through lactation day 21 resulted in dystocia with prolonged parturition and lower offspring body weights at exposures greater than or equal to 11 times the unbound human AUC at the MRHD of 200 mg and lower postnatal survival at exposures equal to 17 times the unbound human AUC at the MRHD of 200 mg. No maternal or developmental toxicity was observed in either dams or offspring at exposures equal to 2.4 times the unbound human AUC at the MRHD of 200 mg.
Administration of Abrocitinib to juvenile rats (comparable to a 3 month old human) resulted in macroscopic and microscopic bone findings. When dosing was initiated at postnatal Day 10 (at exposures greater than or equal to 0.8 times the unbound human AUC at the MRHD of 200 mg), macroscopic bone findings (malrotated and/or impaired use of forelimbs or hindlimbs or paws, fractures, and/or femoral head abnormalities) were noted. Only the microscopic bone dystrophy finding (similar to that observed in rat general toxicity studies of up to 1 month) was fully reversible after cessation of treatment.