Summary of the Safety Profile: The nilotinib safety profile is based on data from patients with newly diagnosed Ph+ CML-CP in a randomized, open label, active comparator-controlled phase-III trial and patients with resistant or intolerant Ph+ CML-CP and CML-AP which served as a basis for the listed indications (see Table 9 and Indications).
In Patients with Newly Diagnosed Ph+ CML-CP: The data reported as follows reflect exposure to Tasigna from a randomized phase III study in patients with newly diagnosed Ph+ CML-CP in chronic phase treated at the recommended dose of 300 mg twice daily (n=279). The median time on treatment was 60.5 months (range 0.1-70.8 months).
Non-hematologic adverse drug reactions (ADRs) reported with very common frequency (≥10%) were rash, pruritus, headache, nausea, fatigue, alopecia, myalgia, and upper abdominal pain. Most of these ADRs were mild to moderate in severity (Grade 1 or 2). Constipation, diarrhea, dry skin, muscle spasms, arthralgia, abdominal pain, peripheral edema, vomiting and asthenia were observed less commonly (<10% and ≥5%) and have been of mild to moderate severity, manageable and generally did not require dose reduction. Pleural and pericardial effusions, regardless of causality, occurred in 2% and <1% of patients, respectively, receiving Tasigna 300 mg twice daily. Gastrointestinal hemorrhage, regardless of causality, was reported in 3% of these patients.
The change from baseline in mean time-averaged QTcF interval at steady-state in the nilotinib recommended dose of 300 mg twice daily was 6 msec. In the nilotinib 400 mg twice daily group and the imatinib 400 mg once daily group the change from baseline in mean time-averaged QTcF interval at steady state were 6 msec and 3 msec, respectively. No patient had an absolute QTcF of >500 msec while on the study drug in any of the Tasigna treatment groups and no events of Torsade de Pointes were observed. QTcF increase from baseline that exceeds 60 msec was observed in 5 patients while on Tasigna (one in the 300 mg twice daily treatment group and four in the 400 mg twice daily treatment group).
No patients in any treatment groups had a LVEF <45% during treatment. Also, there were no patients with 15% or greater decrease from baseline in LVEF.
No sudden deaths have been reported in any treatment group.
In the nilotinib 300 mg twice daily group, hematologic ADRs include myelosuppression: Thrombocytopenia (18%), neutropenia (15%), and anemia (8%). Biochemistry ADRs include alanine aminotransferase increased (24%), hyperbilirubinemia (16%), aspartate aminotransferase increased (12%), lipase increased (11%), blood bilirubin increased (10%), hyperglycemia (4%), hypercholesterolemia (3%), and hypertriglyceridemia (<1%). See Table 9 for Grade 3/4 laboratory abnormalities. (See Table 9.)
Discontinuation due to adverse drug reactions was observed in 10% of patients.
In Patients with Resistant or Intolerant Ph+ CML-CP and CML-AP: The data reported as follows reflect exposure to Tasigna in 458 patients with Ph+ CML-CP (n=321) and CML-AP (n=137) resistant to or intolerant to at least one prior therapy including imatinib in an open-label multicenter study treated at the recommended dose of 400 mg twice daily.
Non-hematologic adverse drug reactions (ADRs) reported with very common frequency (≥10% in the combined CML-CP and CML-AP patient populations) were rash, pruritus, nausea, fatigue, headache, constipation, diarrhea, vomiting and myalgia. Most of these ADRs were mild to moderate in severity. Alopecia, muscle spasms, decreased appetite, arthralgia, bone pain, abdominal pain, peripheral edema and asthenia were observed less frequently (<10% and ≥5%) and have been of mild to moderate severity (Grade 1 or 2).
Pleural and pericardial effusions as well as complications of fluid retention occurred in <1% of patients receiving Tasigna. Cardiac failure was observed in <1% of patients. Gastrointestinal and CNS hemorrhage was reported in 1% and <1% of patients, respectively.
QTcF exceeding 500 msec was observed in this study in 4 patients (<1%). No episodes of Torsade de Pointes (transient or sustained) were observed.
Hematologic ADRs including myelosuppression: Thrombocytopenia (31%), neutropenia (17%) and anemia (14%). See Table 9 for Grade 3/4 laboratory abnormalities. (See Table 9.)
Discontinuation due to adverse drug reactions was observed in 16% of CP and 10% of AP patients.
Most Frequently Reported Adverse Drug Reactions: Non-hematologic ADRs (excluding laboratory abnormalities) that are reported in at least 5% of the patients in any of the Tasigna clinical studies are shown in Table 9. These are ranked under heading of frequency, the most frequent first. Within each frequency grouping adverse drug reactions represented in order of decreasing seriousness. In addition the corresponding frequency category for each adverse drug reaction is based on the following (CIOMS III) convention: Very common (≥1/10) or common (≥1/100 to <1/10). The frequency is based on the highest for any Tasigna group in the two studies, using one decimal precision for percentages. (See Table 9.)
 Click on icon to see table/diagram/image
Additional Data From Clinical Trials:
Click on icon to see table/diagram/image
Additional Data From Clinical Trials: The following adverse drug reactions were reported in patients in the Tasigna clinical studies at the recommended doses at a frequency of less than 5% (common is ≥1/100 to <1/10; uncommon is ≥1/1,000 to <1/100; single events are captured as frequency not known). For laboratory abnormalities, very common events (≥1/10) not included in Table 9 are also reported. These adverse reactions are included based on clinical relevance and ranked in decreasing order of seriousness within each category obtained from two clinical studies: Newly diagnosed Ph+ CML-CP 60 months' analysis and resistant or intolerant Ph+ CML-CP and CML-AP 24 months' analysis.
Infections and Infestations: Common: Folliculitis, upper respiratory tract infection (including pharyngitis, nasopharyngitis, rhinitis). Uncommon: Pneumonia, bronchitis, urinary tract infection, herpes virus infection, candidiasis (including oral candidiasis), gastroenteritis. Frequency Not Known: Sepsis, subcutaneous abscess, anal abscess, furuncle, tinea pedis.
Neoplasms Benign, Malignant and Unspecified: Common: Skin papilloma. Frequency Not Known: Oral papilloma, paraproteinemia.
Blood and Lymphatic System Disorders: Common: Leukopenia, eosinophilia, febrile neutropenia, pancytopenia, lymphopenia. Frequency Not Known: Thrombocythemia, leukocytosis.
Immune System Disorders: Frequency Not Known: Hypersensitivity.
Endocrine Disorders: Uncommon: Hyperthyroidism, hypothyroidism. Frequency Not Known: Hyperparathyroidism secondary, thyroiditis.
Metabolism and Nutrition Disorders: Very Common: Hypophosphatemia (including blood phosphorus decreased). Common: Electrolyte imbalance (including hypomagnesemia, hyperkalemia, hypokalemia, hyponatremia, hypocalcemia, hypercalcemia, hyper-phosphatemia), diabetes mellitus, hyperglycemia, hypercholesterolemia, hyperlipidemia, hypertriglyceridemia. Uncommon: Gout, dehydration, increased appetite, dyslipidemia. Frequency Not Known: Hyperuricemia, hypoglycemia.
Psychiatric Disorders: Common: Depression, insomnia, anxiety. Frequency Not Known: Disorientation, confusional state, amnesia, dysphoria.
Nervous System Disorders: Common: Dizziness, peripheral neuropathy, hypoesthesia, paresthesia. Uncommon: Intracranial hemorrhage, ischemic shock, transient ischemic attack, cerebral infarction, migraine, loss of consciousness (including syncope), tremor, disturbance in attention, hyperesthesia. Frequency Not Known: Cerebrovascular accident, basilar artery stenosis, brain edema, optic neuritis, lethargy, dysesthesia, restless leg syndrome.
Eye Disorders: Common: Eye hemorrhage, periorbital edema, eye pruritus, conjunctivitis, dry eye (including xerophthalmia). Uncommon: Vision impairment, vision blurred, visual acuity reduced, eyelid edema, photopsia, hyperemia (sclera, conjunctival, ocular), eye irritation, conjunctival hemorrhage. Frequency Not Known: Papilloedema, diplopia, photophobia, eye swelling, blepharitis, eye pain, chorioretinopathy, conjunctivitis allergic, ocular surface disease.
Ear and Labyrinth Disorders: Common: Vertigo. Frequency Not Known: Impaired hearing, ear pain, tinnitus.
Cardiac Disorders: Common: Angina pectoris, arrythmia (including atrioventricular block, cardiac flutter, extrasystoles, atrial fibrillation, tachycardia, bradycardia), palpitations, electrocardiogram QT prolonged. Uncommon: Cardiac failure, myocardial infarction, coronary artery disease, cardiac murmur, pericardial effusion, cyanosis. Frequency Not Known: Ventricular dysfunction, pericarditis, ejection fraction decrease.
Vascular Disorders: Common: Hypertension, flushing. Uncommon: Hypertensive crisis, peripheral arterial occlusive disease, intermittent claudication, arterial stenosis limb, hematoma, arteriosclerosis. Frequency Not known: Hemorrhagic shock, hypotension, thrombosis, peripheral artery stenosis.
Respiratory, Thoracic and Mediastinal Disorders: Common: Dyspnea, dyspnea exertional, epistaxis, cough, dysphonia. Uncommon: Pulmonary edema, pleural effusion, interstitial lung disease, pleuritic pain, pleurisy, pharyngolaryngeal pain, throat irritation. Frequency Not Known: Pulmonary hypertension, wheezing, oropharyngeal pain.
Gastrointestinal Disorders: Common: Pancreatitis, abdominal discomfort, abdominal distension, dyspepsia, dysgeusia, flatulence. Uncommon: Gastrointestinal hemorrhage, melena, mouth ulceration, gastroesophageal reflux, stomatitis, esophageal pain, dry mouth, gastritis, sensitivity of teeth. Frequency Not Known: Gastrointestinal ulcer perforation, retroperitoneal hemorrhage, haematemesis, gastric ulcer, ulcerative esophagitis, subileus, enterocolitis, hemorrhoids, hiatus hernia, rectal hemorrhage, gingivitis.
Hepatobiliary Disorders: Very Common: Hyperbilirubinemia (including blood bilirubin increased). Common: Hepatic function abnormal. Uncommon: Hepatotoxicity, toxic hepatitis, jaundice. Frequency Not Known: Cholestasis, hepatomegaly.
Skin and Subcutaneous Tissue Disorders: Common: Night sweats, eczema, urticaria, hyperhidrosis, contusion, acne, dermatitis (including allergic, exfoliative and acneiform). Uncommon: Exfoliative rash, drug eruption, pain of skin, ecchymosis, swelling face. Frequency Not Known: Psoriasis, erythema multiforme, erythema nodosum, skin ulcer, palmar-plantar erythrodysesthesia syndrome, petechiae, photosensitivity, blister, dermal cyst, sebaceous hyperplasia, skin atrophy, skin discoloration, skin exfoliation, skin hyperpigmentation, skin hypertrophy; hyperkeratosis.
Musculoskeletal and Connective Tissue Disorders: Common: Musculoskeletal chest pain, musculoskeletal pain, back pain, neck pain, flank pain, muscle weakness. Uncommon: Musculoskeletal stiffness, joint swelling. Frequency Not Known: Arthritis.
Renal and Urinary Disorders: Common: Pollakiuria. Uncommon: Dysuria, micturition urgency, nocturia. Frequency Not Known: Renal failure, hematuria, urinary incontinence, chromaturia.
Reproductive System and Breast Disorders: Uncommon: Breast pain, gynecomastia, erectile dysfunction. Frequency Not Known: Breast induration, menorrhagia, nipple swelling.
General Disorders and Administration Site Conditions: Common: Pyrexia, chest pain (including non-cardiac chest pain), pain, chest discomfort, malaise. Uncommon: Face edema, gravitational edema, influenza-like illness, chills, feeling body temperature change (including feeling hot, feeling cold). Frequency Not Known: Localized edema.
Investigations: Very Common: Alanine aminotransferase increased, aspartate amino-transferase increased, lipase increased, lipoprotein cholesterol (including low density and high density) increased, total cholesterol increased, blood triglycerides increased. Common: Hemoglobin decreased, blood amylase increased, gamma-glutamyltransferase increased, blood creatinine phosphokinase increased, blood alkaline phosphatase increased, blood insulin increased, weight decreased, weight increased, globulins decreased. Uncommon: Blood lactate dehydrogenase increased, blood urea increased. Frequency Not Known: Troponin increased, blood bilirubin unconjugated increased, blood insulin decreased, insulin C-peptide decreased, blood parathyroid hormone increased.
Laboratory Abnormalities: Clinically relevant or severe abnormalities of routine hematologic or biochemistry laboratory values are presented in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
Adverse Drug Reactions from Spontaneous Reports and Literature Cases (Frequency Not Known): The following adverse reactions have been derived from post marketing experience with Tasigna via spontaneous case reports, literature cases, expanded access programs, and clinical studies other than the global registration trials. Because these reactions are reported from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to nilotinib exposure.
Frequency Not Known: Tumor lysis syndrome.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image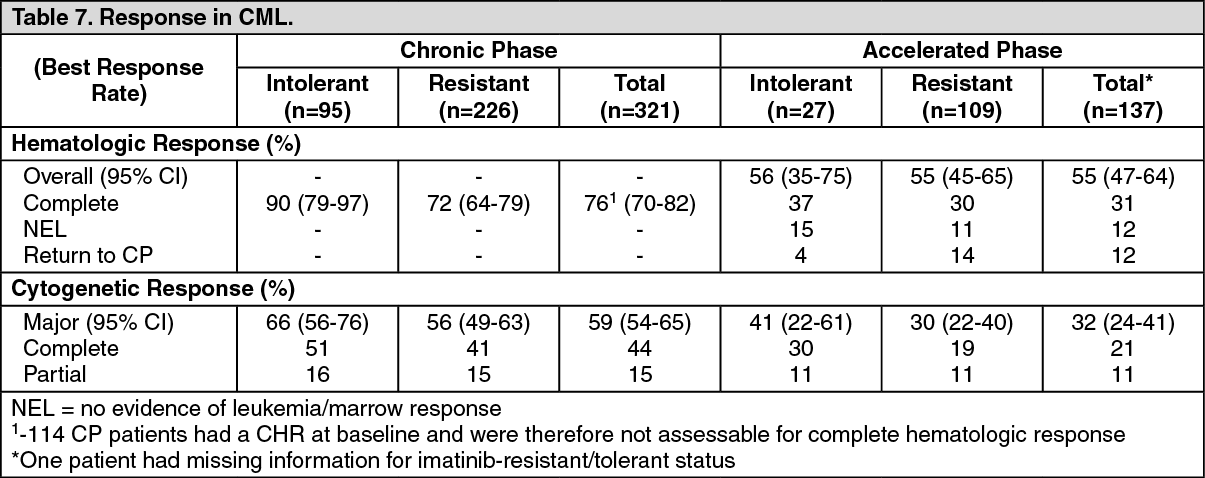 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image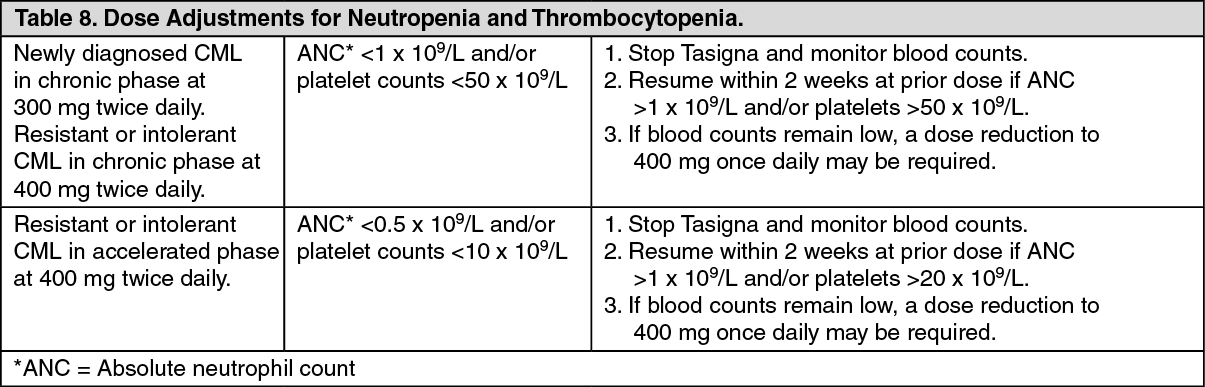 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image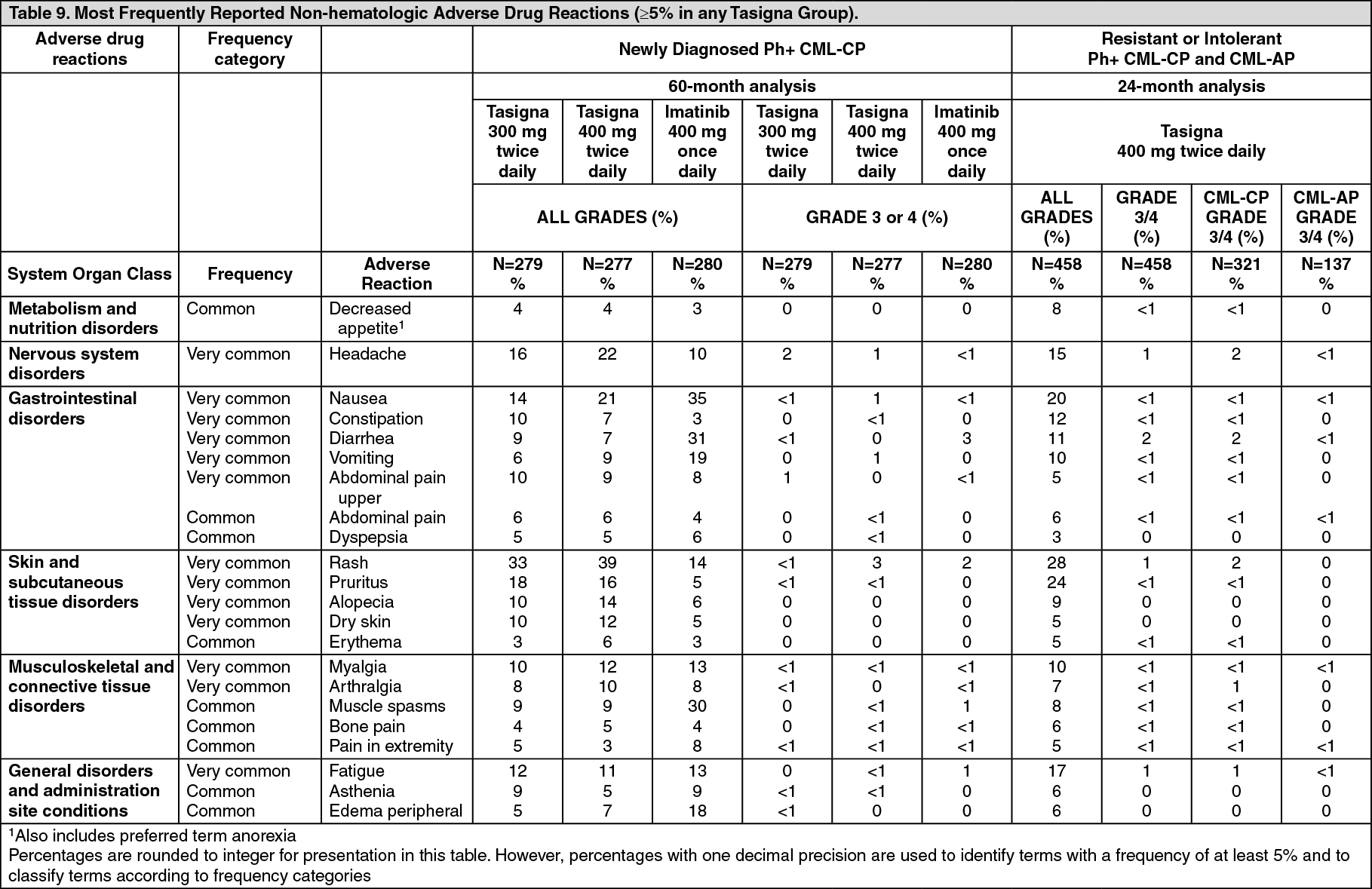 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image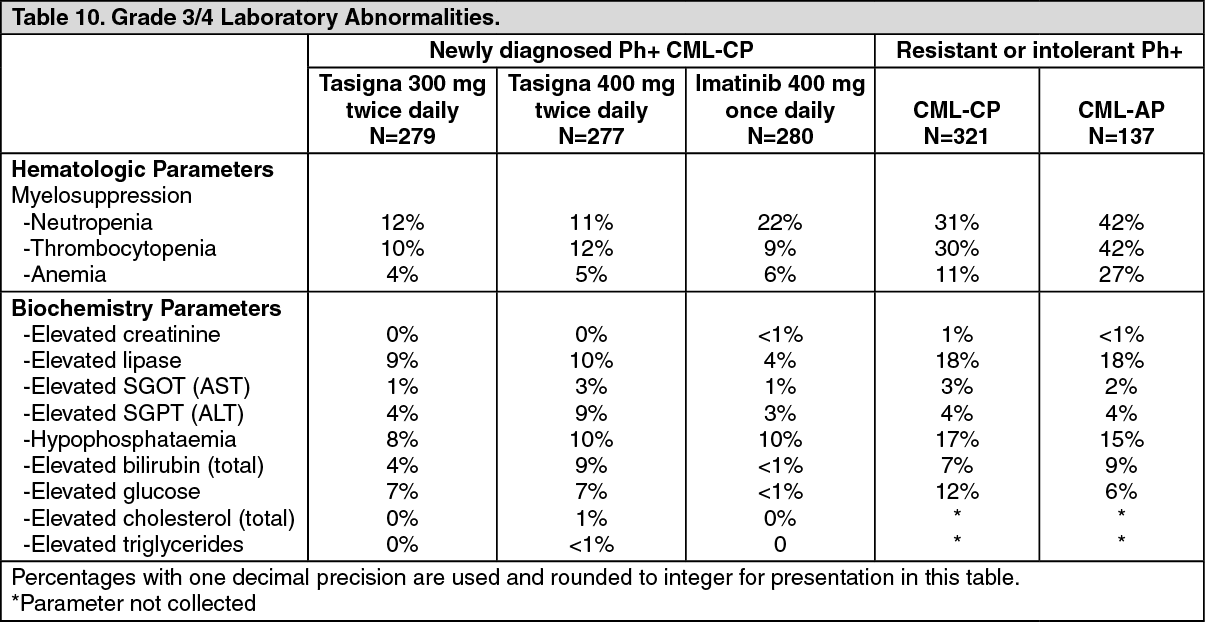 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
