


 Sign Out
Sign Out


Pharmacology: Mechanism of Action: The mechanism of action of aripiprazole, in schizophrenia or bipolar mania, is unknown. However, the efficacy of aripiprazole could be mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonist activity at 5-HT2A receptors. Actions at receptors other than D2, 5-HT1A, and 5-HT2A may explain some of the other clinical effects of aripiprazole (e.g., the orthostatic hypotension observed with aripiprazole may be explained by its antagonist activity at adrenergic alpha1 receptors).
Pharmacodynamics: Aripiprazole exhibits high affinity for dopamine D2 and D3, serotonin 5-HT1A and 5-HT2A receptors (Ki values of 0.34 nM, 0.8 nM, 1.7 nM, and 3.4 nM, respectively), moderate affinity for dopamine D4, serotonin 5-HT2C and 5-HT7, alpha1-adrenergic and histamine H1 receptors (Ki values of 44 nM, 15 nM, 39 nM, 57 nM, and 61 nM, respectively), and moderate affinity for the serotonin reuptake site (Ki=98 nM). Aripiprazole has no appreciable affinity for cholinergic muscarinic receptors (IC50>1000 nM). Aripiprazole functions as a partial agonist at the dopamine D2 and the serotonin 5-HT1A receptors, and as an antagonist at serotonin 5-HT2A receptor.
CLINICAL STUDIES: Schizophrenia: Adults: The efficacy of ABILIFY in the treatment of schizophrenia was evaluated in five short-term (4- and 6-week), placebo-controlled trials of acutely relapsed inpatients who predominantly met DSM-III/IV criteria for schizophrenia. Four of the five trials were able to distinguish ABILIFY from placebo, but one study, the smallest, did not. Three of these studies also included an active control group consisting of either risperidone (one trial) or haloperidol (two trials), but they were not designed to allow for a comparison of ABILIFY and the active comparators.
In the four positive trials for ABILIFY, four primary measures were used for assessing psychiatric signs and symptoms. The Positive and Negative Syndrome Scale (PANSS) is a multi-item inventory of general psychopathology used to evaluate the effects of drug treatment in schizophrenia. The PANSS positive subscale is a subset of items in the PANSS that rates seven positive symptoms of schizophrenia (delusions, conceptual disorganization, hallucinatory behaviour, excitement, grandiosity, suspiciousness/persecution, and hostility). The PANSS negative subscale is a subset of items in the PANSS that rates seven negative symptoms of schizophrenia (blunted affect, emotional withdrawal, poor rapport, passive apathetic withdrawal, difficulty in abstract thinking, lack of spontaneity/flow of conversation, and stereotyped thinking). The Clinical Global Impression (CGI) assessment reflects the impression of a skilled observer, fully familiar with the manifestations of schizophrenia, about the overall clinical state of the patient.
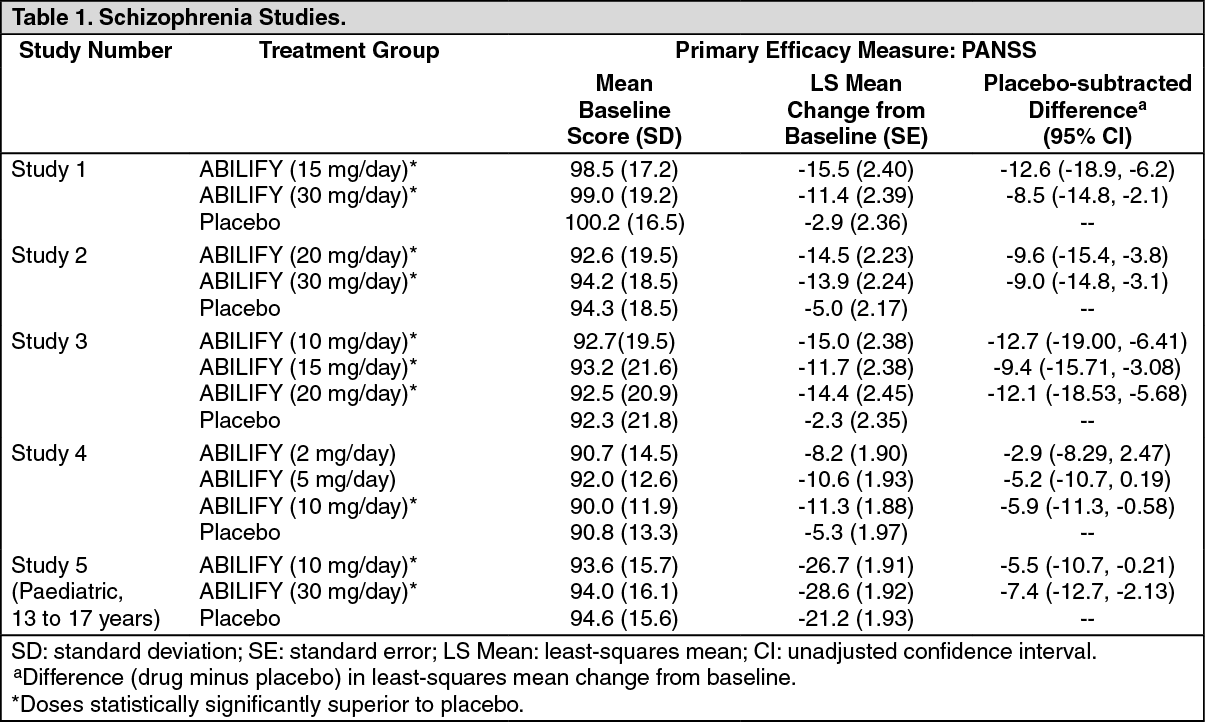
In a 4-week trial (n = 414) comparing two fixed doses of ABILIFY (15 or 30 mg/day) to placebo, both doses of ABILIFY were superior to placebo in the PANSS total score (Study 1 in Table 1), PANSS positive subscale, and CGI-severity score. In addition, the 15-mg dose was superior to placebo in the PANSS negative subscale.
In a 4-week trial (n = 404) comparing two fixed doses of ABILIFY (20 or 30 mg/day) to placebo, both doses of ABILIFY were superior to placebo in the PANSS total score (Study 2 in Table 1), PANSS positive subscale, PANSS negative subscale, and CGI-severity score.
In a 6-week trial (n = 420) comparing three fixed doses of ABILIFY (10, 15, or 20 mg/day) to placebo, all three doses of ABILIFY were superior to placebo in the PANSS total score (Study 3 in Table 1), PANSS positive subscale, and the PANSS negative subscale.
In a 6-week trial (n = 367) comparing three fixed doses of ABILIFY (2, 5, or 10 mg/day) to placebo, the 10-mg dose of ABILIFY was superior to placebo in the PANSS total score (Study 4 in Table 1), the primary outcome measure of the study. The 2-mg and 5-mg doses did not demonstrate superiority to placebo on the primary outcome measure.
Thus, the efficacy of 10-mg, 15-mg, 20-mg, and 30-mg daily doses was established in two studies for each dose. Among these doses, there was no evidence that the higher dose groups offered any advantage over the lowest dose group of these studies. An examination of population subgroups did not reveal any clear evidence of differential responsiveness on the basis of age, gender, or race.
A longer-term trial enrolled 310 inpatients or outpatients meeting DSM-IV criteria for schizophrenia who were, by history, symptomatically stable on other antipsychotic medications for periods of 3 months or longer. These patients were discontinued from their antipsychotic medications and randomized to ABILIFY 15 mg or placebo for up to 26 weeks of observation for relapse. Relapse during the double-blind phase was defined as CGI-Improvement score of ≥ 5 (minimally worse), scores ≥ 5 (moderately severe) on the hostility or uncooperativeness items of the PANSS, or ≥ 20% increase in the PANSS total score. Patients receiving ABILIFY 15 mg experienced a significantly longer time to relapse over the subsequent 26 weeks compared to those receiving placebo.
Paediatric Patients: The efficacy of ABILIFY (aripiprazole) in the treatment of schizophrenia in paediatric patients (13 to 17 years of age) was evaluated in one 6-week, placebo-controlled trial of outpatients who met DSM-IV criteria for schizophrenia and had a PANSS score ≥ 70 at baseline. In this trial (n = 302) comparing two fixed doses of ABILIFY (10 or 30 mg/day) to placebo, ABILIFY was titrated starting from 2 mg/day to the target dose in 5 days in the 10 mg/day treatment arm and in 11 days in the 30 mg/day treatment arm. Both doses of ABILIFY were superior to placebo in the PANSS total score (Study 5 in Table 1), the primary outcome measure of the study. The 30 mg/day dosage was not shown to be more efficacious than the 10 mg/day dose. Although maintenance efficacy in paediatric patients has not been systematically evaluated, maintenance efficacy can be extrapolated from adult data along with comparisons of aripiprazole pharmacokinetic parameters in adult and paediatric patients. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageBipolar Disorder: Acute Treatment of Manic and Mixed Episodes: Adults: Monotherapy: The efficacy of ABILIFY as monotherapy in the acute treatment of manic episodes was established in four 3-week, placebo-controlled trials in hospitalized patients who met the DSM-IV criteria for bipolar I disorder with manic or mixed episodes. These studies included patients with or without psychotic features and two of the studies also included patients with or without a rapid-cycling course.
The primary instrument used for assessing manic symptoms was the Young Mania Rating Scale (Y-MRS), an 11-item clinician-rated scale traditionally used to assess the degree of manic symptomatology in a range from 0 (no manic features) to 60 (maximum score). A key secondary instrument included the Clinical Global Impression-Bipolar (CGI-BP) Scale.
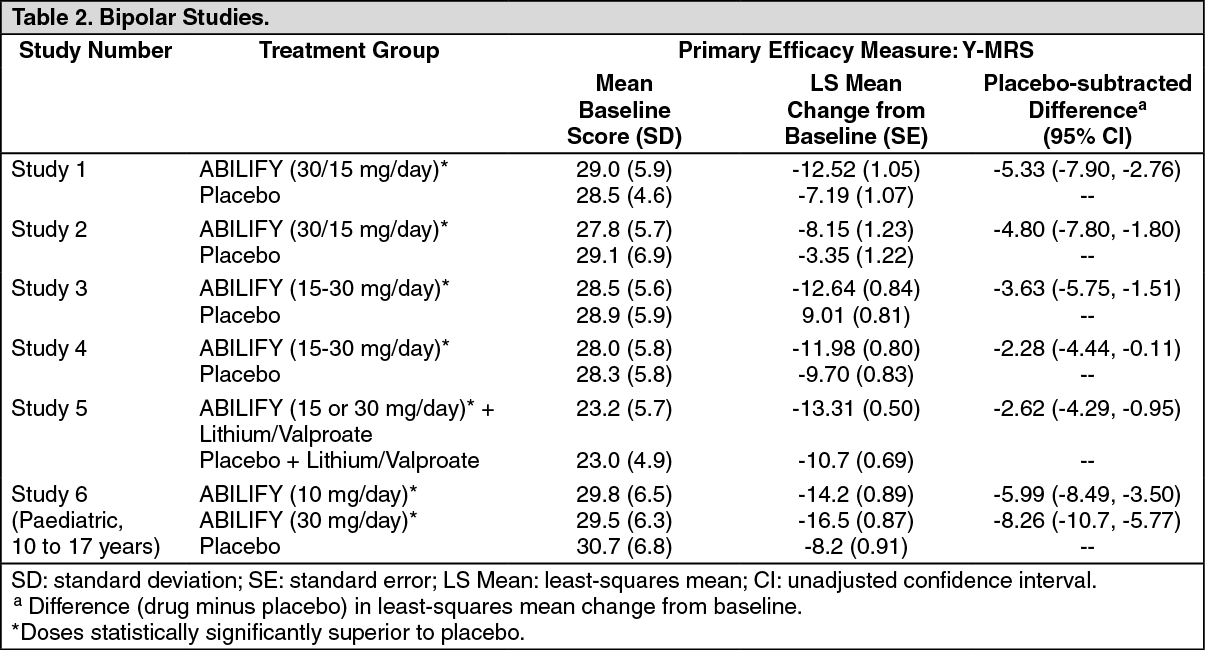
In the four positive, 3-week, placebo-controlled trials (n = 268; n = 248; n = 480; n = 485) which evaluated ABILIFY in a range of 15 mg to 30 mg, once daily (with a starting dose of 30 mg/day in two studies and 15 mg/day in two studies), ABILIFY was superior to placebo in the reduction of Y-MRS total score (Studies 1 to 4 in Table 2) and CGI-BP Severity of Illness score (mania). In the two studies with a starting dose of 15 mg/day, 48% and 44% of patients were on 15 mg/day at endpoint. In the two studies with a starting dose of 30 mg/day, 86% and 85% of patients were on 30 mg/day at endpoint.
Adjunctive Therapy: The efficacy of adjunctive ABILIFY with concomitant lithium or valproate in the treatment of manic or mixed episodes was established in a 6-week, placebo-controlled study (n = 384) with a 2-week lead-in mood stabilizer monotherapy phase in adult patients who met DSM-IV criteria for bipolar I disorder. This study included patients with manic or mixed episodes and with or without psychotic features.
Patients were initiated on open-label lithium (0.6 to 1.0 mEq/L) or valproate (50 to 125 μg/mL) at therapeutic serum levels, and remained on stable doses for 2 weeks. At the end of 2 weeks, patients demonstrating inadequate response (YMRS total score ≥ 16 and ≤ 25% improvement on the Y-MRS total score) to lithium or valproate were randomized to receive either ABILIFY (15 mg/day or an increase to 30 mg/day as early as day 7) or placebo as adjunctive therapy with open-label lithium or valproate. In the 6-week, placebo-controlled phase, adjunctive ABILIFY starting at 15 mg/day with concomitant lithium or valproate (in a therapeutic range of 0.6 to 1.0 mEq/L or 50 to 125 μg/mL, respectively) was superior to lithium or valproate with adjunctive placebo in the reduction of the Y-MRS total score (Study 5 in Table 2) and CGI-BP Severity of Illness score (mania). Seventy-one percent of the patients co-administered valproate and 62% of the patients co-administered lithium were on 15 mg/day at 6-week endpoint.
Paediatric Patients: The efficacy of ABILIFY in the treatment of bipolar I disorder in paediatric patients (10 to 17 years of age) was evaluated in one 4-week, placebo-controlled trial (n=296) of outpatients who met DSM-IV criteria for bipolar I disorder manic or mixed episodes with or without psychotic features and had a Y-MRS score ≥ 20 at baseline. This double-blind, placebo-controlled trial compared two fixed doses of ABILIFY (10 or 30 mg/day) to placebo. The ABILIFY dose was started at 2 mg/day, which was titrated to 5 mg/day after 2 days, and to the target dose in 5 days in the 10 mg/day treatment arm, and in 13 days in the 30 mg/day treatment arm. Both doses of ABILIFY were superior to placebo in change from baseline to week 4 on the Y-MRS total score (Study 6 in Table 2). (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMaintenance Treatment of Bipolar I Disorder: Monotherapy Maintenance Therapy: A maintenance trial was conducted in adult patients meeting DSM-IV criteria for Bipolar I Disorder with a recent manic or mixed episode who had been stabilized on open-label ABILIFY and who had maintained a clinical response for at least 6 weeks. The first phase of this trial was an open-label stabilization period in which inpatients and outpatients were clinically stabilized and then maintained on open-label ABILIFY (15 or 30 mg/day, with a starting dose of 30 mg/day) for at least 6 consecutive weeks. One hundred sixty-one outpatients were then randomized in a double-blind fashion, to either the same dose of ABILIFY they were on at the end of the stabilization and maintenance period or placebo and were then monitored for manic or depressive relapse. During the randomization phase, ABILIFY was superior to placebo on time to the number of combined affective relapses (manic plus depressive), the primary outcome measure for this study. The majority of these relapses were due to manic rather than depressive symptoms. There is insufficient data to know whether ABILIFY is effective in delaying the time to occurrence of depression in patients with Bipolar I Disorder.
An examination of population subgroups did not reveal any clear evidence of differential responsiveness on the basis of age and gender; however, there were insufficient numbers of patients in each of the ethnic groups to adequately assess inter-group differences.
Adjunctive Maintenance Therapy: An adjunctive maintenance trial was conducted in adult patients meeting DSMIV criteria for bipolar I disorder with a recent manic or mixed episode. Patients were initiated on open-label lithium (0.6 to 1.0 mEq/L) or valproate (50 to 125 μg/mL) at therapeutic serum levels, and remained on stable doses for 2 weeks. At the end of 2 weeks, patients demonstrating inadequate response (Y-MRS total score ≥ 16 and ≤ 35% improvement on the Y-MRS total score) to lithium or valproate received ABILIFY with a starting dose of 15 mg/day with the option to increase to 30 mg or reduce to 10 mg as early as day 4, as adjunctive therapy with open-label lithium or valproate. Prior to randomization, patients on the combination of single-blind ABILIFY and lithium or valproate were required to maintain stability (Y-MRS and MADRS total scores ≤ 12) for 12 consecutive weeks. Three hundred thirty-seven patients were then randomized in a double-blind fashion, to either the same dose of ABILIFY they were on at the end of the stabilization period or placebo plus lithium or valproate and were then monitored for manic, mixed, or depressive relapse for a maximum of 52 weeks. ABILIFY was superior to placebo on the primary endpoint, time from randomization to relapse to any mood. A mood event was defined as hospitalization for a manic, mixed, or depressive episode, study discontinuation due to lack of efficacy accompanied by Y-MRS score > 16 and/or a MADRS > 16, or an SAE of worsening disease accompanied by YMRS score > 16 and/or a MADRS > 16. A total of 68 mood events were observed during the double-blind treatment phase. Twenty-five were from the ABILIFY group and 43 were from the placebo group. The number of observed manic episodes in the ABILIFY group (7) were fewer than that in the placebo group (19), while the number of depressive episodes in the ABILIFY group (14) was similar to that in the placebo group (18).
Adjunctive Treatment of Major Depressive Disorder: The efficacy of ABILIFY in the adjunctive treatment of major depressive disorder (MDD) was demonstrated in three short-term (6-week), placebo-controlled trials of adult patients meeting DSM-IV criteria for MDD who had an inadequate response to prior antidepressant therapy (1 to 3 courses) in the current episode and who had also demonstrated an inadequate response to 8 weeks of prospective antidepressant therapy (paroxetine controlled-release, venlafaxine extended-release, fluoxetine, escitalopram, or sertraline). Inadequate response for prospective treatment was defined as less than 50% improvement on the 17-item version of the Hamilton Depression Rating Scale (HAMD17), minimal HAMD17 score of 14, and a Clinical Global Impressions Improvement rating of no better than minimal improvement. Inadequate response to prior treatment was defined as less than 50% improvement as perceived by the patient after a minimum of 6 weeks of antidepressant therapy at or above the minimal effective dose.
The primary instrument used for assessing depressive symptoms was the Montgomery-Asberg Depression Rating Scale (MADRS), a 10-item clinician-rated scale used to assess the degree of depressive symptomatology. The key secondary instrument was the Sheehan Disability Scale (SDS), a 3-item self-rated instrument used to assess the impact of depression on three domains of functioning with each item scored from 0 (not at all) to 10 (extreme).
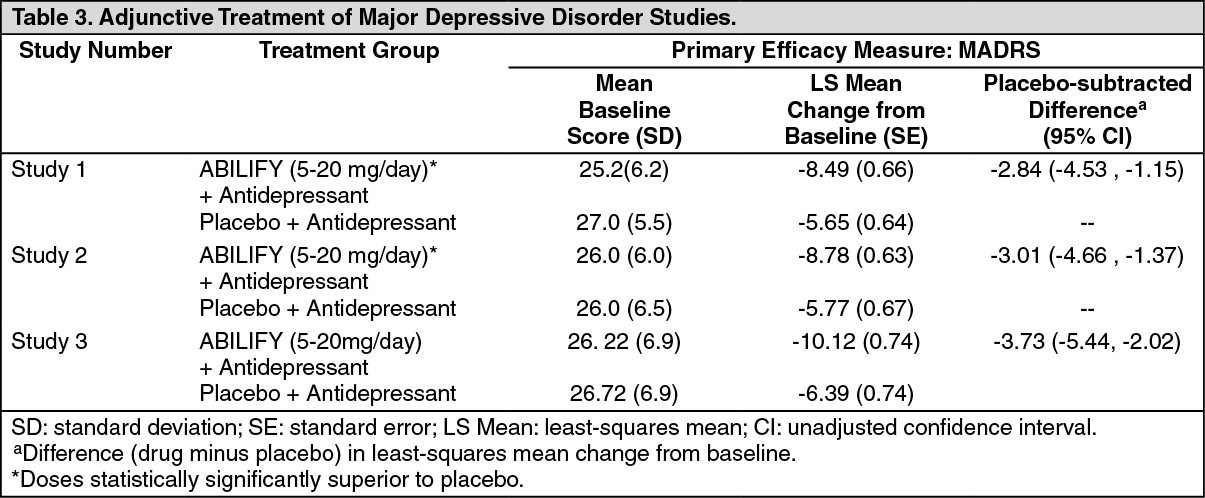
In the three trials (n = 381, n = 362, n = 349), ABILIFY was superior to placebo in reducing mean MADRS total scores (Studies 1, 2, and 3 in Table 3). In one study, ABILIFY was also superior to placebo in reducing the mean SDS score.
In these trials, patients received ABILIFY adjunctive to antidepressants at a dose of 5 mg/day. Based on tolerability and efficacy, doses could be adjusted by 5 mg increments, one week apart. Allowable doses were: 2, 5, 10, 15 mg/day, and for patients who were not on potent CYP2D6 inhibitors fluoxetine and paroxetine, 20 mg/day. All patients who tolerated the initial dose were titrated to at least 10 mg/day. The mean final dose at the end point for the three trials was 10.7, 11.4, and 10.7 mg/day.
An examination of population subgroups did not reveal evidence of differential response based on age, choice of prospective antidepressant, or race. With regard to gender, a smaller mean reduction on the MADRS total score was seen in males than in females.
The statistical significance of the treatment-by-gender effect in the combined analysis is primarily due to the results of one of 3 studies. Consistent results between males and females were reported in 2 of the 3 studies. The clinical relevance of this apparent treatment-by-gender is unknown.
Clinical trials evaluating ABILIFY in MDD did not include ABILIFY monotherapy treatment arms. It is therefore unknown whether efficacy in adjunct treatment is due to ABILIFY alone or from combined treatment with an ADT. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIrritability Associated with Autistic Disorder: Paediatric Patients: The efficacy of ABILIFY (aripiprazole) in the treatment of irritability associated with autistic disorder was established in two 8-week, placebo-controlled trials in paediatric patients (6 to 17 years of age) who met the DSM-IV criteria for autistic disorder and demonstrated behaviours such as tantrums, aggression, self-injurious behaviour, or a combination of these problems. These patients had clinically significant behavioural problems that were at least moderate in severity, as defined by a CGI-Severity score ≥ 4 and an Irritability Subscale score ≥ 18 on the Aberrant Behaviour Checklist (ABC). Over 75% of these subjects were under 13 years of age.
Efficacy was evaluated using two assessment scales: the ABC and the Clinical Global Impression-Improvement (CGI-I) scale. The primary outcome measure in both trials was the change from baseline to endpoint in the Irritability subscale of the ABC (ABC-I). The ABC-I subscale measured symptoms of irritability in autistic disorder.
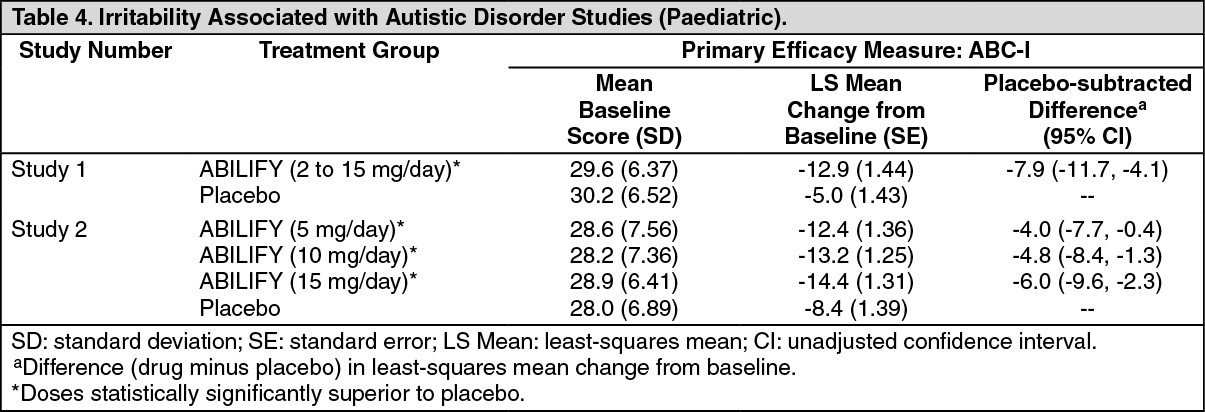
The results of these trials are as follows: In one of the 8-week, placebo-controlled trials, children and adolescents with autistic disorder (n = 98), aged 6 to 17 years, received daily doses of placebo or ABILIFY 2 to 15 mg/day. ABILIFY, starting at 2 mg/day with increases allowed up to 15 mg/day based on clinical response, significantly improved scores on the ABC-I subscale and on the CGI-I scale compared with placebo. The mean daily dose of ABILIFY at the end of 8week treatment was 8.6 mg/day (Study 1 in Table 4).
In the other 8-week, placebo-controlled trial in children and adolescents with autistic disorder (n = 218), aged 6 to 17 years, three fixed doses of ABILIFY (5 mg/day, 10 mg/day, or 15 mg/day) were compared to placebo. ABILIFY dosing started at 2 mg/day and was increased to 5 mg/day after one week. After a second week, it was increased to 10 mg/day for patients in the 10 and 15 mg dose arms, and after a third week, it was increased to 15 mg/day in the 15 mg/day treatment arm (Study 2 in Table 4). All three doses of ABILIFY significantly improved scores on the ABC-I subscale compared with placebo. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn a study designed to assess safety and efficacy of Aripiprazole in the long-term maintenance treatment of paediatric patients (6 to 17 years of age) with irritability associated with autistic disorder, the efficacy of ABILIFY for the maintenance treatment of irritability associated with autistic disorder was not established.
This study was a multicentre, double-blind, randomized, placebo-controlled study with 2 parallel treatment groups designed to assess the safety and efficacy of aripiprazole in the long-term maintenance treatment of paediatric subjects with irritability associated with autistic disorder. The study included 2 phases: Phase 1 (stabilization phase) - 13 to 26 weeks of single-blind aripiprazole treatment and Phase 2 (randomization phase) - 16 weeks of double-blind treatment with aripiprazole or placebo.
The primary objective was to evaluate the efficacy of aripiprazole compared with placebo to prevent relapses in paediatric subjects who maintained a response for 12 weeks of aripiprazole treatment for their symptoms of irritability associated with autistic disorder as measured by the time from randomization to relapse.
This study did not meet its primary end point. The Kaplan-Meier relapse rates at Week 16 were 32% for aripiprazole and 50% for placebo (p = 0.097; hazard ratio of 0.57; 95% CI: 0.28, 1.12). The effect size observed was smaller than what was used to power the study.
Tourette's Disorder: Paediatric Patients: The efficacy of ABILIFY (aripiprazole) in the treatment of Tourette's disorder was established in one 8-week (7 to 17 years of age) and one 10-week (6 to 18 years of age), placebo-controlled trials in paediatric patients (6 to 18 years of age) who met the DSM-IV criteria for Tourette's disorder and had a Total Tic score (TTS) ≥ 20 - 22 on the Yale Global Tic Severity Scale (YGTSS). The YGTSS is a fully validated scale designed to measure current tic severity.
Efficacy was evaluated using two assessment scales: 1) the Total Tic score (TTS) of the YGTSS and 2) the Clinical Global Impressions Scale for Tourette's Syndrome (CGI-TS), a clinician-determined summary measure that takes into account all available patient information. Over 65% of these patients were under 13 years of age.
The primary outcome measure in both trials was the change from baseline to endpoint in the TTS of the YGTSS. Ratings for the TTS are made along 5 different dimensions on a scale of 0 to 5 for motor and vocal tics each. Summation of these 10 scores provides a TTS (i.e., 0 to 50).
The results of these trials are as follows: In the 8-week, placebo-controlled, fixed-dose trial, children and adolescents with Tourette's disorder (n = 133), aged 7 to 17 years, were randomized 1:1:1 to low dose ABILIFY, high dose ABILIFY, or placebo. The target doses for the low and high dose ABILIFY groups were based on weight. Patients < 50 kg in the low dose ABILIFY group started at 2 mg per day with a target dose of 5 mg per day after 2 days. Patients ≥ 50 kg in the low dose ABILIFY group, started at 2 mg per day increased to 5 mg per day after 2 days, with a subsequent increase to a target dose of 10 mg per day at day 7. Patients < 50 kg in the high dose ABILIFY group started at 2 mg per day increased to 5 mg per day after 2 days, with a subsequent increase to a target dose of 10 mg per day at day 7. Patients ≥ 50 kg in the high dose ABILIFY group, started at 2 mg per day increased to 5 mg per day after 2 days, with a subsequent increase to a dose of 10 mg per day at day 7 and were allowed weekly increases of 5 mg per day up to a target dose 20 mg per day at Day 21. ABILIFY (both high and low dose groups) demonstrated statistically significantly improved scores on the YGTSS TTS (Study 1 in Table 5) and on the CGI-TS scale compared with placebo. The estimated improvements on the YGTSS TTS over the course of the study are displayed in figure. (See figure.)
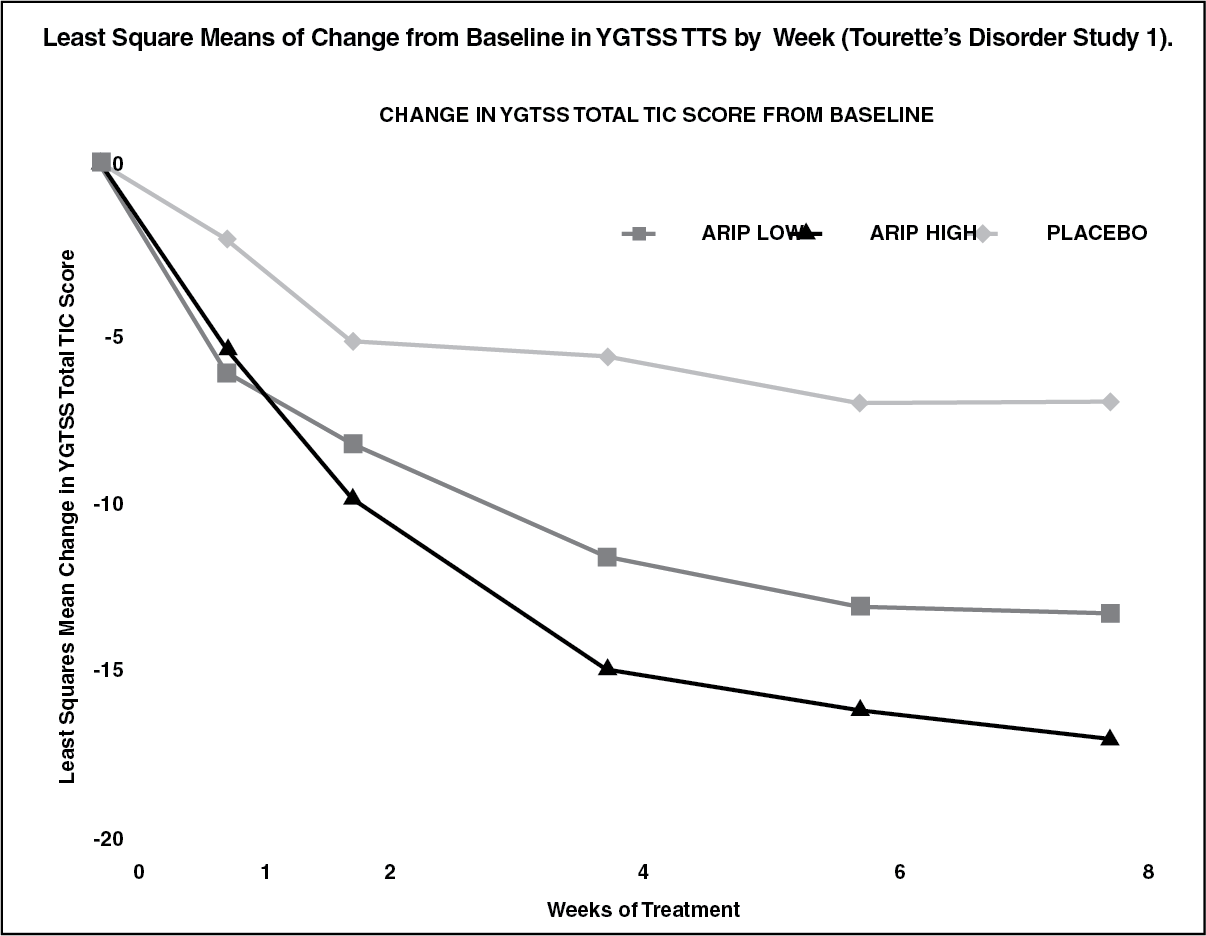 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the 10-week, placebo-controlled, flexible-dose trial in children and adolescents with Tourette's disorder (n = 61), aged 6 to 18 years, patients received daily doses of placebo or ABILIFY, starting at 2 mg/day with increases allowed up to 20 mg/day based on clinical response. ABILIFY demonstrated statistically significantly improved scores on the YGTSS TTS scale compared with placebo (Study 2 in Table 5). The mean daily dose of ABILIFY at the end of 10-week treatment was 6.54 mg/day. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: ABILIFY activity is presumably primarily due to the parent drug, aripiprazole, and to a lesser extent, to its major metabolite, dehydro-aripiprazole, which has been shown to have affinities for D2 receptors similar to the parent drug and represents 40% of the parent drug exposure in plasma. The mean elimination half-lives are about 75 hours and 94 hours for aripiprazole and dehydro-aripiprazole, respectively. Steady-state concentrations are attained within 14 days of dosing for both active moieties. Aripiprazole accumulation is predictable from single-dose pharmacokinetics. At steady state, the pharmacokinetics of aripiprazole is dose-proportional. Elimination of aripiprazole is mainly through hepatic metabolism involving two P450 isozymes, CYP2D6 and CYP3A4. For CYP2D6 poor metabolizers, the mean elimination half-life for aripiprazole is about 146 hours.
Absorption: Tablet: Aripiprazole is well absorbed after administration of the tablet, with peak plasma concentrations occurring within 3 hours to 5 hours; the absolute oral bioavailability of the tablet formulation is 87%. ABILIFY can be administered with or without food. Administration of a 15-mg ABILIFY tablet with a standard high-fat meal did not significantly affect the Cmax or AUC of aripiprazole or its active metabolite, dehydro-aripiprazole, but delayed Tmax by 3 hours for aripiprazole and 12 hours for dehydro-aripiprazole.
Oral Solution: Aripiprazole is well absorbed when administered orally as the solution. At equivalent doses, the plasma concentrations of aripiprazole from the solution were higher than that from the tablet formulation. In a relative bioavailability study comparing the pharmacokinetics of 30 mg aripiprazole as the oral solution to 30 mg aripiprazole tablets in healthy subjects, the solution to tablet ratios of geometric mean Cmax and AUC values were 122% and 114%, respectively [see Dosing of Oral Solution under DOSAGE & ADMINISTRATION]. The single-dose pharmacokinetics of aripiprazole were linear and dose-proportional between the doses of 5 mg to 30 mg
Distribution: The steady-state volume of distribution of aripiprazole following intravenous administration is high (404 L or 4.9 L/kg), indicating extensive extravascular distribution. At therapeutic concentrations, aripiprazole and its major metabolite are greater than 99% bound to serum proteins, primarily to albumin. In healthy human volunteers administered 0.5 to 30 mg/day aripiprazole for 14 days, there was dose-dependent D2-receptor occupancy indicating brain penetration of aripiprazole in humans.
Metabolism and Elimination: Aripiprazole is metabolized primarily by three biotransformation pathways: dehydrogenation, hydroxylation, and N-dealkylation. Based on in vitro studies, CYP3A4 and CYP2D6 enzymes are responsible for dehydrogenation and hydroxylation of aripiprazole, and N-dealkylation is catalysed by CYP3A4. Aripiprazole is the predominant drug moiety in the systemic circulation. At steady state, dehydro-aripiprazole, the active metabolite, represents about 40% of aripiprazole AUC in plasma.
Following a single oral dose of [14C]-labelled aripiprazole, approximately 25% and 55% of the administered radioactivity was recovered in the urine and faeces, respectively. Less than 1% of unchanged aripiprazole was excreted in the urine and approximately 18% of the oral dose was recovered unchanged in the faeces.
Toxicology: NONCLINICAL TOXICOLOGY: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenesis: Lifetime carcinogenicity studies were conducted in ICR mice, Sprague-Dawley (SD) and F344 rats. Aripiprazole was administered for 2 years in the diet at doses of 1, 3, 10, and 30 mg/kg/day to ICR mice and 1, 3, and 10 mg/kg/day to F344 rats (0.2 to 5 times and 0.3 to 3 times the maximum recommended human dose [MRHD] based on mg/m2, respectively). In addition, SD rats were dosed orally for 2 years at 10, 20, 40, and 60 mg/kg/day (3 to 19 times the MRHD based on mg/m2). Aripiprazole did not induce tumours in male mice or male rats. In female mice, the incidences of pituitary gland adenomas and mammary gland adenocarcinomas and adenoacanthomas were increased at dietary doses of 3 to 30 mg/kg/day (0.1 to 0.9 times human exposure at MRHD based on AUC and 0.5 to 5 times the MRHD based on mg/m2). In female rats, the incidence of mammary gland fibro adenomas was increased at a dietary dose of 10 mg/kg/day (0.1 times human exposure at MRHD based on AUC and 3 times the MRHD based on mg/m2); and the incidences of adrenocortical carcinomas and combined adrenocortical adenomas/carcinomas were increased at an oral dose of 60 mg/kg/day (14 times human exposure at MRHD based on AUC and 19 times the MRHD based on mg/m2).
Proliferative changes in the pituitary and mammary gland of rodents have been observed following chronic administration of other antipsychotic agents and are considered prolactin-mediated. Serum prolactin was not measured in the aripiprazole carcinogenicity studies. However, increases in serum prolactin levels were observed in female mice in a 13-week dietary study at the doses associated with mammary gland and pituitary tumours. Serum prolactin was not increased in female rats in 4-week and 13-week dietary studies at the dose associated with mammary gland tumours. The relevance for human risk of the findings of prolactin-mediated endocrine tumours in rodents is unknown.
Mutagenesis: The mutagenic potential of aripiprazole was tested in the in vitro bacterial reverse-mutation assay, the in vitro bacterial DNA repair assay, the in vitro forward gene mutation assay in mouse lymphoma cells, the in vitro chromosomal aberration assay in Chinese hamster lung (CHL) cells, the in vivo micronucleus assay in mice, and the unscheduled DNA synthesis assay in rats. Aripiprazole and a metabolite (2,3-DCPP) were clastogenic in the in vitro chromosomal aberration assay in CHL cells with and without metabolic activation. The metabolite, 2,3-DCPP, produced increases in numerical aberrations in the in vitro assay in CHL cells in the absence of metabolic activation. A positive response was obtained in the in vivo micronucleus assay in mice; however, the response was shown to be due to a mechanism not considered relevant to humans.
Impairment of Fertility: Female rats were treated with oral doses of 2, 6, and 20 mg/kg/day (0.6, 2, and 6 times the maximum recommended human dose [MRHD] on a mg/m2 basis) of aripiprazole from 2 weeks prior to mating through day 7 of gestation. Oestrus cycle irregularities and increased corpora lutea were seen at all doses, but no impairment of fertility was seen. Increased pre-implantation loss was seen at 6 and 20 mg/kg/day, and decreased foetal weight was seen at 20 mg/kg/day.
Male rats were treated with oral doses of 20, 40, and 60 mg/kg/day (6, 13, and 19 times the MRHD on a mg/m2 basis) of aripiprazole from 9 weeks prior to mating through mating. Disturbances in spermatogenesis were seen at 60 mg/kg, and prostate atrophy was seen at 40 and 60 mg/kg, but no impairment of fertility was seen.
Animal Toxicology and/or Pharmacology: Aripiprazole produced retinal degeneration in albino rats in a 26-week chronic toxicity study at a dose of 60 mg/kg and in a 2-year carcinogenicity study at doses of 40 and 60 mg/kg. The 40- and 60-mg/kg/day doses are 13 and 19 times the maximum recommended human dose (MRHD) based on mg/m2 and 7 to 14 times human exposure at MRHD based on AUC. Evaluation of the retinas of albino mice and of monkeys did not reveal evidence of retinal degeneration. Additional studies to further evaluate the mechanism have not been performed. The relevance of this finding to human risk is unknown.