


 Sign Out
Sign Out
Based on the data from clinical trials, exposure-response relationships for any Grade 3 or 4 adverse reaction were observed at higher exposures, with a faster time to onset for adverse reactions with increasing pralsetinib exposure.
Dose reductions due to adverse reactions occurred in 41.5% of patients treated with Gavreto. The most common adverse reactions resulting in dose reductions were neutropenia (14.0%), anaemia (8.5%), lymphopenia (5.3%), pneumonitis (5.3%), leukopenia (4.2%), blood creatine phosphokinase increased (4.0%), hypertension (4.0%), and fatigue (3.8%).
Permanent discontinuation due to adverse reactions occurred in 8.1% of patients treated with Gavreto. The most common adverse reactions that led to permanent discontinuation of Gavreto were pneumonia and pneumonitis (1.9% for each).
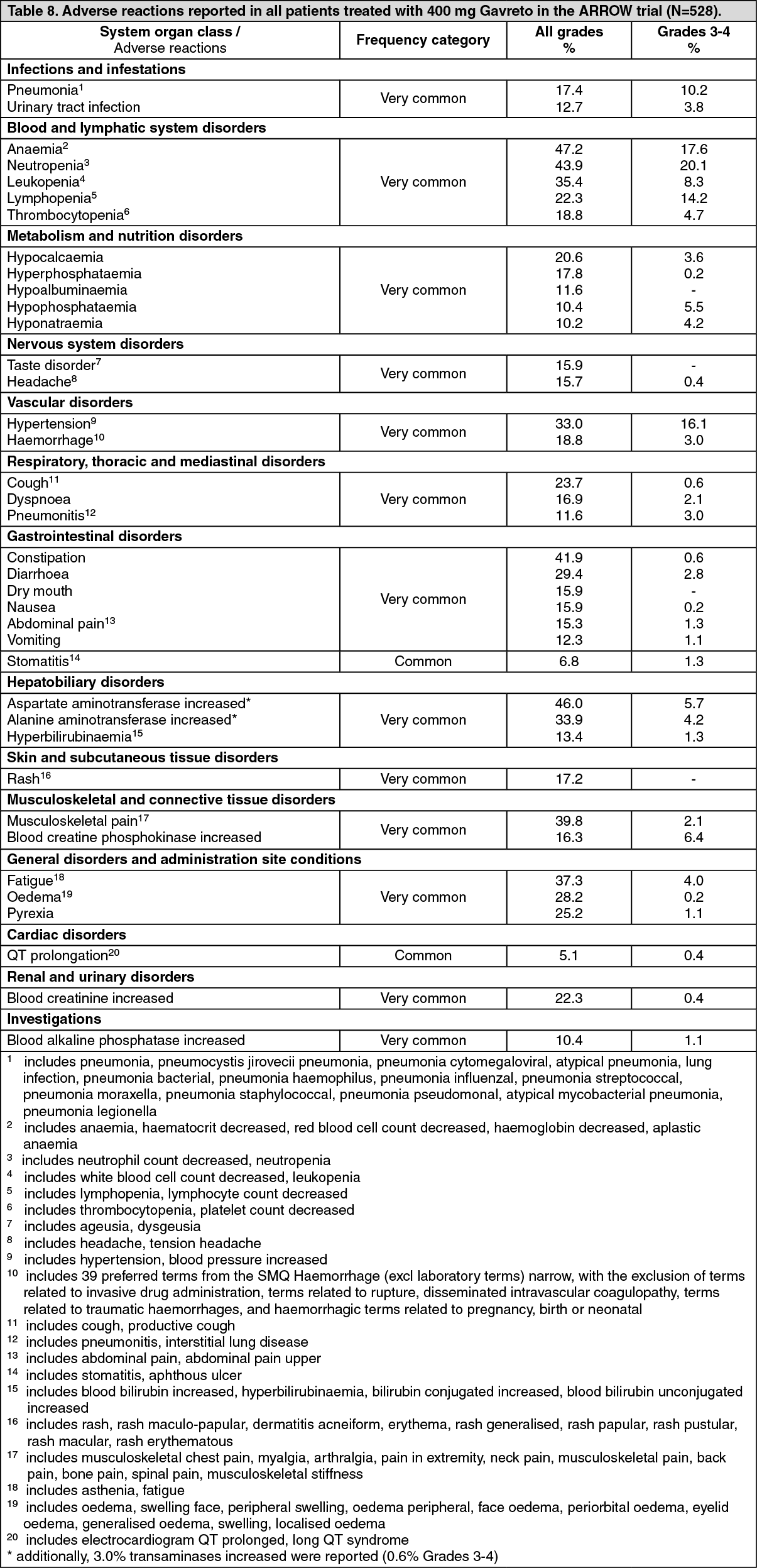
Tabulated list of adverse reactions: The safety population includes a total of 528 patients, including 281 patients with advanced NCSLC, as well as patients with other solid tumours (including RET fusion thyroid cancer and RET mutation medullary thyroid cancer), who received pralsetinib at a starting dose of 400 mg, see PHARMACOLOGY: Pharmacodynamics under Actions. No clinically relevant differences in the safety profile across indications have been observed.
Adverse reactions reported in patients treated with Gavreto in the ARROW trial are listed as follows (Table 8), according to the MedDRA System Organ Class and frequency.
Frequencies are defined using the following convention: very common (≥1/10); common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), and not known (cannot be estimated from the available data).
Within each system organ class, adverse reactions are presented in order of decreasing frequency and severity. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Pneumonitis/ILD: Pneumonitis and ILD occurred in 11.6% of 528 patients with NSCLC or other solid tumours, enrolled in the ARROW Study who received Gavreto (see Precautions). Among the patients who had pneumonitis/ILD, the median time to onset was 15.6 weeks.
Serious adverse reactions of pneumonitis/ILD were reported for 5.3% of patients, including Grade 3 events (2.5%), Grade 4 (0.6%) and one fatal (Grade 5) event (0.2%).
In clinical trials, the majority of the patients with Grade 1 or Grade 2 pneumonitis were able to continue treatment without recurrent pneumonitis/ILD following dose interruption and dose reduction. Dose interruption occurred in 8.9%, dose reduction in 5.3% and permanent dose discontinuation in 1.9% of patients due to ILD/pneumonitis. The median time to resolution was 3.7 weeks.
Hypertension: Hypertension (including blood pressure increased) occurred in 33.0% of 528 patients with NSCLC or other solid tumours, including Grade ≤2 events in 16.9% and Grade 3 in 16.1% of patients. No Grade 4 or Grade 5 events were reported. Among the patients who had hypertension, the median time to onset was 2.1 weeks.
Serious adverse reactions of hypertension were reported in 1.3% of all patients (all Grade 3 events).
Dose interruption occurred in 7.4% of patients, dose reduction in 4.0% and one patient (0.2%) required permanent dose discontinuation. The median time to resolution was 3.1 weeks.
Transaminase elevations: Increased AST occurred in 46.0% of 528 patients, including Grade 3 or 4 in 5.7% of patients. Increased ALT occurred in 33.9% of patients, including Grade 3 or 4 events in 4.2% of patients. The median time to first onset for increased AST was 2.1 weeks and increased ALT was 3.1 weeks.
Serious adverse reactions of increased AST and ALT were each reported for 0.6% of all patients.
Dose interruption due to increased AST or ALT occurred in 4.4% and 3.4% of patients, respectively and dose reduction in 1.3% for both events. No patients required permanent dose discontinuation. The median time to resolution was 5.3 and 4.1 weeks for increased AST and ALT, respectively.
Haemorrhagic events: Haemorrhagic events occurred in 18.8% of the 528 patients, including Grade 3 events in 2.8% of patients and a Grade 4 or fatal (Grade 5) event each occurred in one patient (0.2%).
Serious adverse reactions of haemorrhage were reported for 3.2% of patients.
Fourteen patients (2.7%) required dose interruption and dose reduction or permanent dose discontinuation due to haemorrhage each occurred in one patient.
QT prolongation: QT prolongation occurred in 5.1% of 528 patients with NSCLC or other solid tumours. In 2 patients (0.4%) the event was assessed as serious. The majority of patients experienced non-severe events - i.e. Grade 1, in 21 (4.0%) and Grade 2, in 4 patients (0.8%). Two patients (0.4%) experienced Grade 3 events of Electrocardiogram QT prolonged, which both resolved. There was no life-threatening or fatal QT prolongation. Three patients (0.6%) had an event that remained unresolved by time of data cut-off. Dose reductions or interruptions were required by two Electrocardiogram QT prolonged patients, each. No QT prolongation event led to permanent discontinuation of pralsetinib.
Infections: Infections were commonly experienced by 57.2% of 528 patients during the median treatment time of 9.5 months. Most frequently (>10%), the preferred terms of pneumonia and urinary tract infection were reported (14.2% and 12.7%, respectively). The majority of infections were mild (Grade 1 or 2) and resolved; severe infection (Grade ≥3) occurred in 23.5% patients (with fatal events reported for 1.9%).
Infections reported as serious occurred for 24.2% of patients. The most common (>2%) serious infection preferred term was pneumonia (9.8%), followed by urinary tract infection (3.4%) and sepsis (2.8%). The majority of patients experiencing sepsis had concurrent pneumonia or urinary tract infection reported.
Dose interruption due to infection occurred for 19.5% of patients (mainly due to the preferred terms of pneumonia [6.8%] and urinary tract infection [2.7%]). Dose was reduced due to infections in 3.2% of patients (mainly due to the preferred term of pneumonia [1.9%]). Permanent treatment discontinuation was required by 3.4% of patients due to infections (mainly due to the preferred term of pneumonia [1.7%]).
Elderly: In ARROW (N=528), 37.8% of patients were 65 years of age and older. Compared with younger patients (<65), more patients of ≥65 years old reported adverse reactions that led to permanent dose discontinuation (25.8% versus 13.4%). Of the commonly reported events with higher incidence in elderly patients (≥65), hypertension has the greatest difference in comparison with patients <65 years of age. However, hypertension is also expected to occur more frequently in the elderly population. Older patients reported more Grade 3 or higher adverse reactions compared to younger patients (87.1% versus 72.3%).
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via Roche Thailand Local Safety Unit at thailand.drug_safety@roche.com.
View ADR Monitoring Form