


 Sign Out
Sign Out
PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Pralsetinib is a tyrosine kinase inhibitor that targets oncogenic RET fusions and mutations, including V804 gatekeeper mutations associated with resistance to other therapies. In vitro, pralsetinib inhibited several oncogenic RET fusions and mutations (CCDC6 RET, RET V804L, RET V804M and RET M918T) with half maximal inhibitory concentrations at clinically relevant concentrations. In a broad panel of purified enzyme assays, pralsetinib demonstrated selectivity for RET with 81-fold selectivity over VEGFR2.
RET fusion proteins and activating point mutations can drive tumorigenic potential through hyperactivation of downstream signaling pathways leading to uncontrolled cell proliferation. Pralsetinib exhibited anti-tumor activity in cultured cells and animal tumor implantation models representing multiple tumor types harboring oncogenic RET fusions or mutations (KIF5B-RET, CCDC6-RET, RET M918T, RET C634W, as well as the V804L and V804M mutants associated with cabozantinib and vandetanib resistance).
Pharmacodynamic effects: Cardiac electrophysiology: The QT interval prolongation potential of pralsetinib was assessed in 34 patients with RET fusion-positive solid tumours administered at 400 mg once daily in a formal ECG sub-study.
In patients receiving pralsetinib in the ARROW study, QT prolongation was reported (see Adverse Reactions). Therefore, dose interruption or modification may be required in patients treated with pralsetinib (see Dosage & Administration and Precautions).
Clinical efficacy and safety: The efficacy of Gavreto was demonstrated in a multi-center, open-label clinical trial in adults (ARROW). Patients with RET-fusion positive NSCLC, thyroid cancer, and other RET-altered advanced solid tumors were included in the study. Identification of a RET gene alteration was determined by local laboratories using next generation sequencing (NGS), fluorescence in situ hybridization (FISH), and other tests.
All NSCLC patients were required to have locally advanced or metastatic disease with measurable disease by Response Evaluable Criteria in Solid Tumours (RECIST) version 1.1. (v1.1) and have a RET fusion as determined by local testing (Next Generation Sequencing (NGS), fluorescence in situ hybridization (FISH), other). Patients with asymptomatic central nervous system (CNS) metastases, including patients with stable or decreasing steroid use within 2 weeks prior to study entry, were enrolled. The protocol excluded patients with a known primary driver alteration other than RET fusions, patients with a history of prolonged QT syndrome or Torsades de pointes or a familial history of prolonged QT syndrome, clinically symptomatic pneumonitis, and any prior or ongoing clinically significant medical condition that could affect patient's safety.
The primary efficacy outcome measure was overall response rate (ORR) according to RECIST v1.1 as evaluated by a Blinded Independent Central Review (BICR). Secondary efficacy outcomes included duration of response (DOR), progression free survival (PFS) and overall survival (OS).
Overall RET fusion-positive NSCLC population: The efficacy population consisted of 233 patients with RET fusion-positive advanced NSCLC who were treated at a starting dose of 400 mg orally once daily, including 75 who were treatment-naïve and 136 who previously received platinum-based chemotherapy. As of the last data cut-off date, the median follow-up was 17.1 months.
The demographic characteristics across the 233 patients were: 52.4% female, 51.9% White, 39.5% Asian, 3.9% Hispanic/Latino, and the median age was 60.0 years (range: 26 to 87) with 37.8% ≥65 years of age. The majority of patients had an ECOG performance status at baseline of 0 (33.5%) or 1 (63.9%), had metastatic disease (97.4%), had never smoked (62.2%) or were former smokers (33.5%) and had adenocarcinoma (96.1%). A history of brain metastases was seen in 37.3% of patients. Patients previously treated with platinum-based chemotherapy (N=136), received a median of 2 prior lines of therapy (range: 1-8). In addition to platinum-based chemotherapy, 40.4% received PD-1/PD-L1 inhibitors, 27.9% received multikinase inhibitors (MKIs) and 47.8% received prior radiation therapy. 21.3% of systemic treatment-naïve patients (N=75) received prior radiation therapy. RET fusions were detected in 79.4% of patients using NGS (42.9% tumour samples; 15.9% blood or plasma samples, 20.6% unknown), 18.0% using FISH, and 2.6% using other methods. The most common RET fusion partners were KIF5B (70.4%) and CCD6 (17.6%).
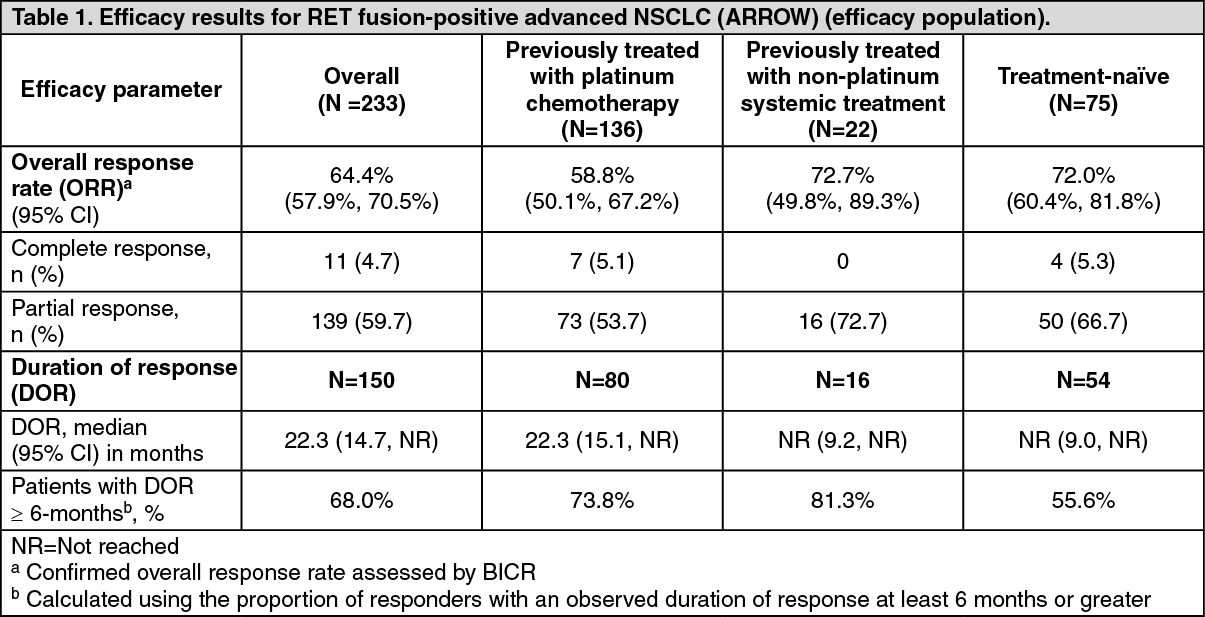
Efficacy results are summarised in Table 1. The median time to first response was 1.8 months for the overall population (range: 0.9-11.4 months), as well as for patients previously treated with platinum chemotherapy (range: 1.3-11.4 months) and treatment-naïve patients (range: 0.9-6.1 months). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageORR and median DOR for the 233 patients with RET fusion-positive advanced NSCLC in the efficacy population was 64.4% (95% CI: 57.9, 70.5) and 22.3 months (95% CI: 14.7, NR), respectively.
No clinically relevant difference in efficacy was seen in patients with a KIF5B or CCDC6 fusion partner. BICR response rates were: ORR=67.7% (95% CI: 59.9, 74.8) in 164 patients with a KIF5B fusion partner; and ORR=68.3% (95% CI: 51.9, 81.9) in 41 patients with a CCDC6 fusion partner.
The intracranial ORR assessed by BICR was 70.0% (95% CI: 34.8, 93.3) in 10 response evaluable patients with brain metastases at baseline, including 3 patients with a complete response. All patients had target brain lesion shrinkage with pralsetinib treatment.
Elderly population: In ARROW (N=528), 37.8% of patients were 65 years of age and older. No overall differences in pharmacokinetic, safety or efficacy were observed in comparison with younger patients.
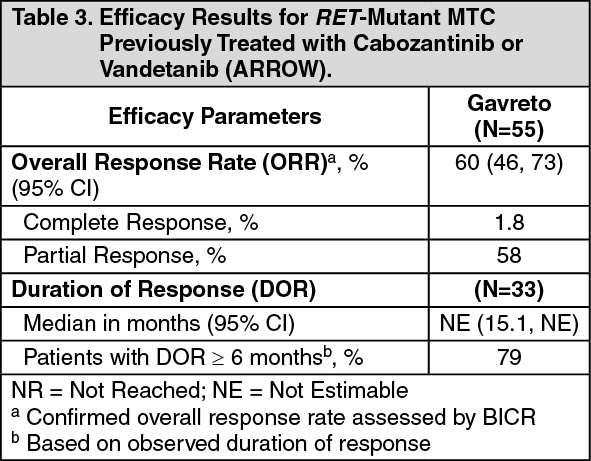
RET-Mutant MTC Previously Treated with Cabozantinib or Vandetanib: Efficacy was evaluated in 55 patients with RET-mutant metastatic MTC previously treated with cabozantinib or vandetanib (or both).
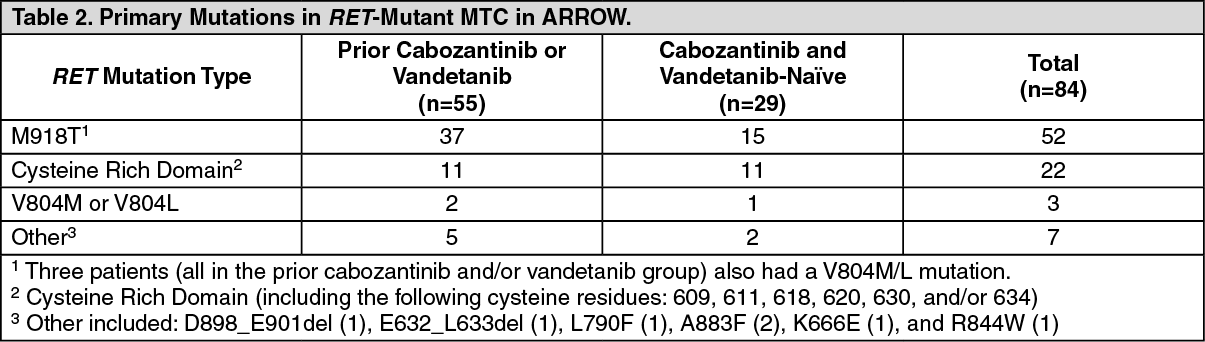
The median age was 59 years (range: 25 to 83); 69% were male, 78% were White, 5% were Asian, 5% were Hispanic/Latino. ECOG performance status was 0-1 (95%) or 2 (5%), and 7% had a history of CNS metastases. Patients had received a median of 2 prior therapies (range 1-7). RET mutation status was detected in 73% using NGS [55% tumor sample, 18% plasma], 26% using PCR sequencing, and 2% other. The primary mutations in RET-mutant MTC previously treated with cabozantinib or vandetanib are described in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEfficacy results for RET-mutant MTC are summarized in Table 3. (See Table 3.)
 Click on icon to see table/diagram/image
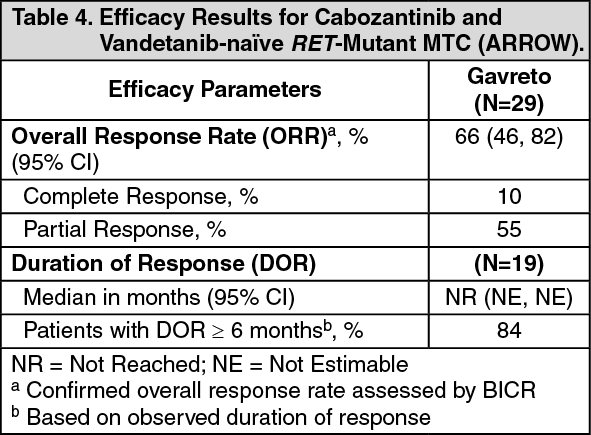
Click on icon to see table/diagram/imageCabozantinib and Vandetanib-naïve RET-mutant MTC: Efficacy was evaluated in 29 patients with RET-mutant advanced MTC who were cabozantinib and vandetanib treatment-naïve.
The median age was 61 years (range: 19 to 81); 72% were male, 76% were White, 17% were Asian, 3.4% were Hispanic/Latino. ECOG performance status was 0-1 (100%), 97% had metastatic disease, and 14% had a history of CNS metastases. Twenty-eight percent (28%) had received up to 3 lines of prior systemic therapy (including 10% PD-1/PD-L1 inhibitors, 10% radioactive iodine, 3.4% kinase inhibitors). RET mutation status was detected in 90% using NGS [52% tumor sample, 35% plasma, 3.4% blood] and 10% using PCR sequencing. The primary mutations used to identify and enroll patients are described in Table 2.
Efficacy results for cabozantinib and vandetanib-naïve RET-mutant MTC are summarized in Table 4. (See Table 4.)
 Click on icon to see table/diagram/image
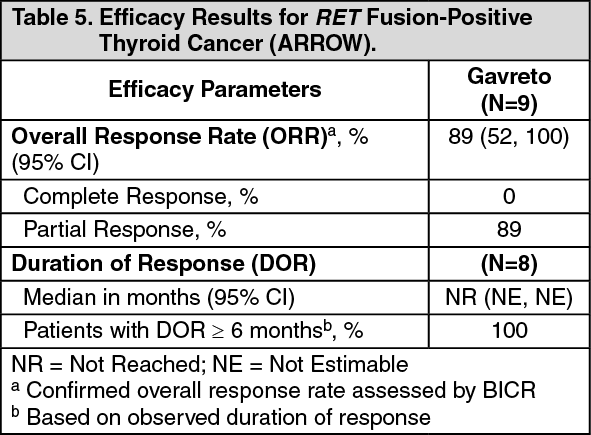
Click on icon to see table/diagram/imageRET Fusion-Positive Thyroid Cancer: The efficacy of GAVRETO was evaluated in RET fusion-positive metastatic thyroid cancer patients in a multicenter, open-label, multi-cohort clinical trial (ARROW, NCT03037385). All patients with RET fusion-positive thyroid cancer were required to have disease progression following standard therapy, measurable disease by RECIST version 1.1, and have RET fusion status as detected by local testing (89% NGS tumor samples and 11% using FISH).
The median age was 61 years (range: 46 to 74); 67% were male, 78% were White, 22% were Asian, 11% were Hispanic/Latino. All patients (100%) had papillary thyroid cancer. ECOG performance status was 0-1 (100%), all patients (100%) had metastatic disease, and 56% had a history of CNS metastases. Patients had received a median of 2 prior therapies (range 1-8). Prior systemic treatments included prior radioactive iodine (100%) and prior sorafenib and/or lenvatinib (56%).
Efficacy results are summarized in Table 5. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Pralsetinib Cmax and AUC increased inconsistently over the dose range of 60 mg to 600 mg once daily (0.15 to 1.5 times the recommended dose); pharmacokinetics was linear in the dose range of 200 and 400 mg in healthy volunteers. Pralsetinib plasma concentrations reached steady state by 3 to 5 days.
At the recommended dose of 400 mg once daily under fasting conditions, the mean steady state Cmax of pralsetinib was 2830 ng/mL and the mean steady state area under the concentration-time curve (AUC0-24h) was 43900 h·ng/mL. The mean accumulation ratio was ~2-fold after repeated dosing.
Absorption: The median time to peak concentration (Tmax) ranged from 2.0 to 4.0 hours following single doses of pralsetinib 60 mg to 600 mg (0.15 to 1.5 times the approved recommended dose). The absolute bioavailability of pralsetinib has not been determined.
Effect of food: Following administration of a single dose of 200 mg Gavreto with a high-fat meal (approximately 800 to 1000 calories with 50 to 60% of calories from fat), the mean (90% CI) Cmax of pralsetinib was increased by 104% (65%, 153%), the mean (90% CI) AUC0-∞ was increased by 122% (96%, 152%), and the median Tmax was delayed from 4 to 8.5 hours, compared to the fasted state.
Distribution: The mean apparent volume of distribution of pralsetinib is 3.8 L/kg (268 L). Plasma protein binding of pralsetinib is 97.1% and is independent of concentration. The blood-to-plasma ratio is 0.6 to 0.7.
Biotransformation: Pralsetinib is primarily metabolised by CYP3A4 and UGT1A4, and to a lesser extent by CYP2D6 and CYP1A2 in vitro.
Following a single oral dose of approximately 310 mg of radiolabelled pralsetinib to healthy subjects, pralsetinib metabolites from oxidation (M531, M453, M549b) and glucuronidation (M709) were detected in small to trace amounts (~ 5%).
Elimination: The mean plasma elimination half-life of pralsetinib was 14.7 hours following a single dose of 400 mg (the recommended dose) pralsetinib and 22.2 hours following multiple doses of 400 mg pralsetinib.
The steady state mean apparent oral clearance of pralsetinib (CL/F) is 9.1 L/h.
Following a single oral dose of radiolabelled pralsetinib to healthy subjects, 72.5% of the radioactive dose was recovered in faeces (66% as unchanged) and 6.1% in urine (4.8% as unchanged).
Interactions with CYP substrates: In vitro studies indicate that pralsetinib is a time-dependent inhibitor of CYP3A4/5 at clinically relevant concentrations. Pralsetinib may have the potential to inhibit or induce CYP2C8, CYP2C9, and CYP3A4/5 at clinically relevant concentrations.
Interactions with transport proteins: In vitro studies indicate that pralsetinib may have the potential to inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, MATE1, and MATE2-K at clinically relevant concentrations. Pralsetinib is a substrate of P-gp (see Interactions).
In vitro studies with drug transporters: In vitro studies indicate that pralsetinib may be a potential substrate of P-glycoprotein (P-gp) and BCRP at clinically relevant concentrations.
Special populations: No clinically relevant differences in the pharmacokinetics of pralsetinib were observed based on age (19 to 87 years), sex, race (White, Black, or Asian), body weight (34.9 to 128 kg), mild to moderate (CLCR 30 to 89 mL/min estimated by Cockcroft-Gault) renal impairment, or mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin > 1 to 1.5 times ULN and any AST). The effect of severe renal impairment (CLCR 15 to 29 mL/min), end-stage renal disease (CLCR < 15 mL/min), or moderate to severe hepatic impairment (total bilirubin > 1.5 times ULN and any AST) on the pharmacokinetics of pralsetinib is unknown (see Dosage & Administration). Hence, no dose modifications are needed in the previously mentioned special populations.
Toxicology: Preclinical safety data: Repeat-dose toxicity studies: In studies of up to 13 weeks duration in rats and cynomolgus monkeys, the primary findings at exposures similar to steady state human exposures (AUC) at 400 mg once daily in patients with advanced NSCLC included physeal dysplasia in the rat (2 times margin) and haematological effects (1 times margin) in both species. Additional adverse findings at higher exposures include degenerative changes in male and female reproductive organs (2 times margin) and increases in blood phosphorus with corresponding mineralization in soft tissues in rats (≥2 times margin), and myocardial haemorrhage in rats (4.4 times margin). Increased blood pressure was observed in rats after a single dose of 25 mg/kg (2 times). The No-Observed-Adverse-Effect-Level (NOAEL) of pralsetinib in the 13-week studies was 10 mg/kg/day in both species, corresponding to exposure (AUC) margins of 1 times relative to the human exposures.
Regarding local exposure and toxicity, there was no evidence of gastrointestinal disturbance in either species up to the NOAEL dose of 10 mg/kg (0.9 times human margin). At higher doses in monkeys, gastrointestinal ulcerations and haemorrhage were observed.
Embryotoxicity/Teratogenicity: In an embryo-fetal development study, administration of pralsetinib to rats during the period of organogenesis was teratogenic and embryotoxic at exposures below the steady-state human clinical exposure (AUC) at 400 mg once daily dose. Malformations, including visceral (primarily kidney and ureter) and skeletal (vertebral, rib, costal cartilage, and vertebral central anomalies) were observed at approximately 0.2-fold of the human exposure. Postimplantation loss occurred at 0.5-fold of the human exposure, and increased to 100% incidence at 1.5-fold of human exposure.
Reproductive toxicity: In a dedicated fertility and early embryonic development study conducted in treated male rats mated to treated female rats pralsetinib did not have an effect on male or female mating performance or ability to become pregnant. However, consistent with the findings of the embryofetal development toxicology study there was post-implantation loss at doses as low as 5 mg/kg (approximately 0.3 times the human exposure (AUC) at the clinical dose of 400 mg based on toxicokinetic data from the 13-week rat toxicology study). At the 20 mg/kg dose level (approximately 2.5-3.6 times the human exposure) 82% of female rats had totally resorbed litters, with 92% post-implantation loss (early resorptions).
In a 13-week repeat-dose toxicology study, male rats exhibited microscopic evidence of tubular degeneration/atrophy in the testis with secondary cellular debris and reduced sperm in the lumen of the epididymis, which correlated with lower mean testis and epididymis weights and gross observations of soft and small testis. Female rats exhibited degeneration of the corpus luteum in the ovary. For both sexes, these effects were observed at pralsetinib doses ≥10 mg/kg/day, approximately 0.9 times the human exposure based on AUC at the clinical dose of 400 mg.
No findings were noted in the reproductive organs in a 13-week repeated-dose toxicology study in monkeys at dose levels up to 10 mg/kg/day (approximately 1 times the human exposure at the 400 mg once daily dose).
Genotoxicity and carcinogenicity: Pralsetinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay and was negative in both in vitro human lymphocyte chromosome aberration assay and in vivo rat bone marrow micronucleus tests.
Carcinogenicity studies with pralsetinib have not been conducted.