


 Sign Out
Sign Out
Three patients (0.8%) in the pooled phase III clinical trials receiving HEMLIBRA prophylaxis withdrew from treatment due to ADRs, which were thrombotic microangiopathy, skin necrosis contemporaneous with superficial thrombophlebitis, and headache.
Adverse drug reactions from the pooled phase III clinical trials in patients who received HEMLIBRA are listed by MedDRA system organ class (see Table 16 as follows). The corresponding frequency categories for each ADR are based on the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), and uncommon (≥ 1/1,000 to < 1/100). (See Table 16.)
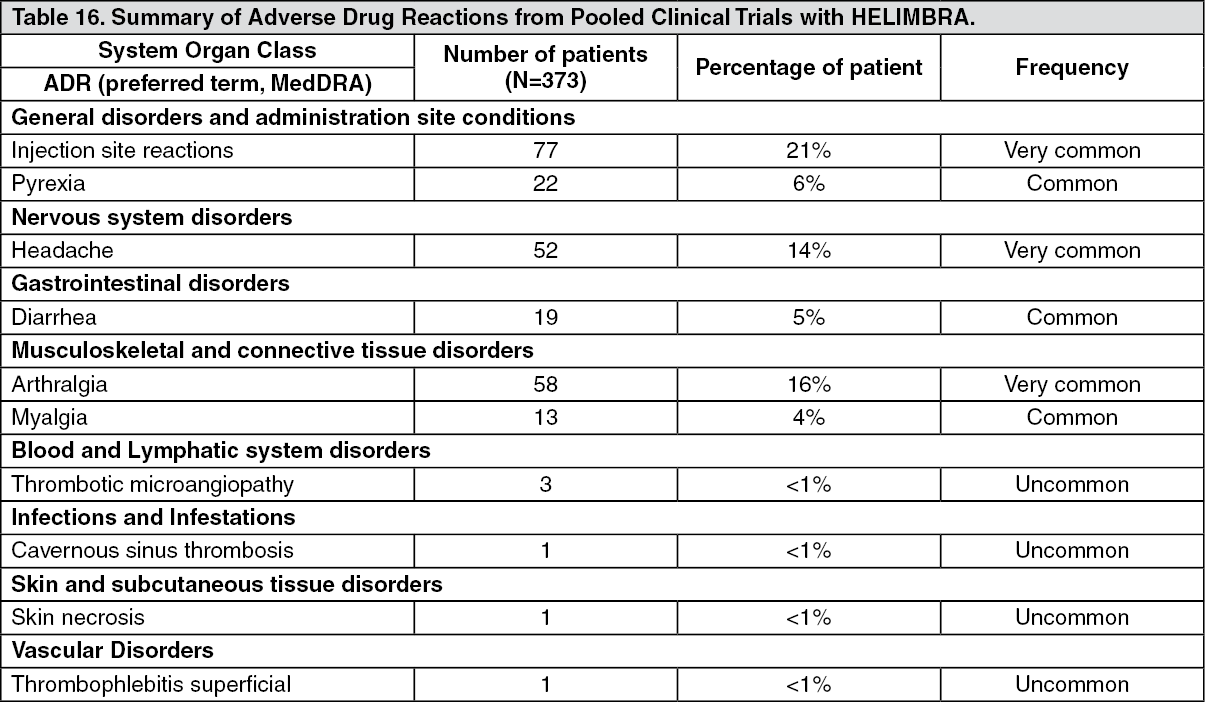 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse drug reactions: The most serious adverse drug reactions reported from the pooled phase III clinical trials with HEMLIBRA were TMA and thrombotic events, including cavernous sinus thrombosis and superficial vein thrombosis contemporaneous with skin necrosis (see as follows and Precautions).
Thrombotic microangiopathy: In the pooled phase III clinical trials, thrombotic microangiopathy events were reported in <1% of patients (3/373) and in 9.7% of patients (3/31) who received at least one dose of aPCC. Each patient was reported to have received on average a cumulative amount of > 100 U/kg/24 hours of aPCC for 24 hours or more while receiving HEMLIBRA prophylaxis prior to the development of TMA events (presenting with thrombocytopenia, microangiopathic hemolytic anemia and acute kidney injury, without severe deficiencies in ADAMTS13 activity). One patient resumed HEMLIBRA following resolution of TMA without recurrence (see Precautions).
Thrombotic events: In the pooled phase III clinical trials, serious thrombotic events were reported in <1% of patients (2/373) and in 6.5% of patients (2/31) who received at least one dose of aPCC. Each patient was reported to have received on average a cumulative amount of > 100 U/kg/24 hours of aPCC for 24 hours or more while receiving HEMLIBRA prophylaxis, prior to the development of the thrombotic events. One patient resumed HEMLIBRA following resolution of the thrombotic event without recurrence (see Precautions).
Characterization of aPCC Treatment (in the pooled phase III clinical trials): There were 82 instances of aPCC treatment*, of which 8 instances (10%) consisted of on average a cumulative amount of > 100 U/kg/24 hours of aPCC for 24 hours or more; two of the 8 instances were associated with thrombotic events and three of the 8 instances were associated with TMA (see Table 17). No TMA or thrombotic events were associated with the remaining instances of aPCC treatment. Of all instances of aPCC treatment, 68% consisted of a single infusion ≤ 100 U/kg. (See Table 17.)
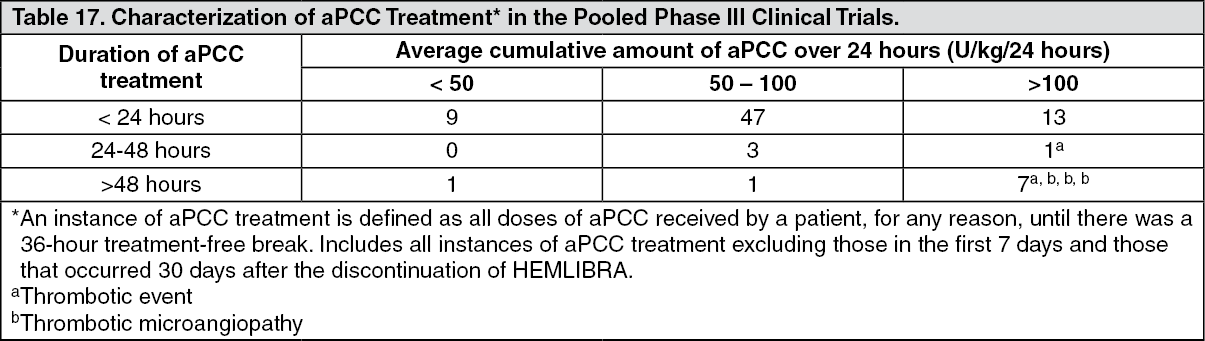 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageInjection site reactions: Injection site reactions (ISRs) were reported very commonly (21%) from clinical trials. All ISRs observed in the HEMLIBRA clinical trials were reported as being non-serious and mild to moderate in intensity, and 95% resolved without treatment. The most commonly reported ISR symptoms were injection site erythema (11%); injection site pain (4%) and injection site pruritus (3%).
Immunogenicity: In the pooled phase III clinical trials with HEMLIBRA, development of neutralizing anti-emicizumab antibodies associated with decreasing emicizumab concentration was uncommon (see PHARMACOLOGY: PHARMACODYNAMICS: IMMUNOGENICITY under Actions). One patient, who developed neutralizing anti-emicizumab antibodies with decreasing emicizumab concentration, experienced loss of efficacy (manifest as breakthrough bleeding) after 5 weeks of treatment and later discontinued HEMLIBRA treatment (see Precautions and PHARMACOLOGY: PHARMACODYNAMICS: IMMUNOGENICITY under Actions). Overall, the safety profile of HEMLIBRA was similar between those patients with anti-emicizumab antibodies (including neutralizing antibodies) and those without.
POST MARKETING: The following adverse drug reactions have been identified from post-marking surveillance with HEMLIBRA (see Table 18). Adverse drug reactions from post-marking surveillance are listed by MedDRA system organ class. (See Table 18.)
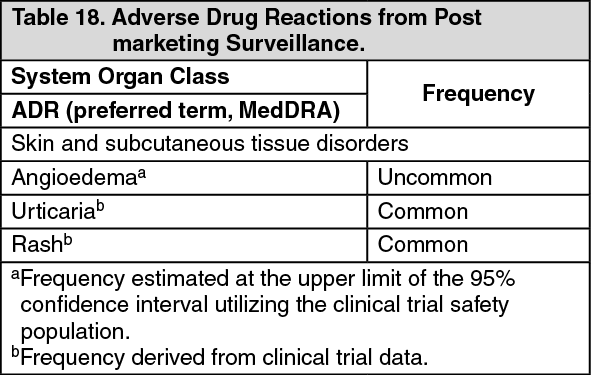 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
View ADR Monitoring Form