


 Sign Out
Sign Out
PHARMACOLOGY: PHARMACODYNAMICS: MECHANISM OF ACTION: Emicizumab bridges activated factor IX and factor X to restore the function of missing activated factor VIII that is needed for effective hemostasis.
Emicizumab has no structural relationship or sequence homology to FVIII and, as such, does not induce or enhance the development of direct inhibitors to FVIII.
Pharmacodynamics: Hemophilia A is an X-linked hereditary disorder of blood coagulation due to a deficiency of functional FVIII and results in bleeding into joints, muscles or internal organs, either spontaneously or as result of accidental or surgical trauma. Prophylactic therapy with HEMLIBRA shortens the aPTT and increases the reported FVIII activity (using a chromogenic assay with human coagulation factors). These two pharmacodynamic markers do not reflect the true hemostatic effect of emicizumab in vivo (aPTT is overly shortened and reported FVIII activity may be overestimated) but provide a relative indication of the pro-coagulant effect of emicizumab.
CLINICAL/EFFICACY STUDIES: The efficacy of HEMLIBRA for routine prophylaxis in patients with hemophilia A with or without inhibitors to FVIII was evaluated in four clinical studies (three adult and adolescent studies [HAVEN 3, HAVEN 1, and HAVEN 4] and a pediatric study [HAVEN 2]).
Clinical Studies in Adult and Adolescent Patients: HAVEN 3: The HAVEN 3 study was a randomized, multicenter, open-label, phase III clinical study in 152 adult and adolescent males (aged ≥ 12 years and ≥ 40 kg) with hemophilia A without FVIII inhibitors who previously received either episodic ("on demand") or prophylactic treatment with FVIII. Patients received subcutaneous HEMLIBRA, 3 mg/kg once weekly for the first four weeks followed by either 1.5 mg/kg once weekly (Arms A and D) or 3 mg/kg every two weeks (Arm B) thereafter, or no prophylaxis (Arm C). Patients in Arm C could switch to HEMLIBRA (3 mg/kg every two weeks) after completing at least 24 weeks without prophylaxis. For Arms A and B dose up-titration to 3 mg/kg weekly was allowed after 24 weeks for patients who experienced two or more qualified bleeds (i.e., spontaneous and clinically significant bleeds occurring at steady state). Arm D patients could up-titrate after the second qualifying bleed. At the time of the analysis, five patients underwent up-titration of their maintenance dose.
Eighty-nine patients previously treated with episodic ("on demand") FVIII were randomized in a 2:2:1 ratio to receive HEMLIBRA either once weekly (Arm A; N = 36), every two weeks (Arm B; N = 35) or no prophylaxis (Arm C; N = 18), with stratification by prior 24-week bleed rate (< 9 or ≥ 9). Sixty-three patients previously treated with prophylactic FVIII were enrolled into Arm D to receive HEMLIBRA (1.5 mg/kg once weekly).
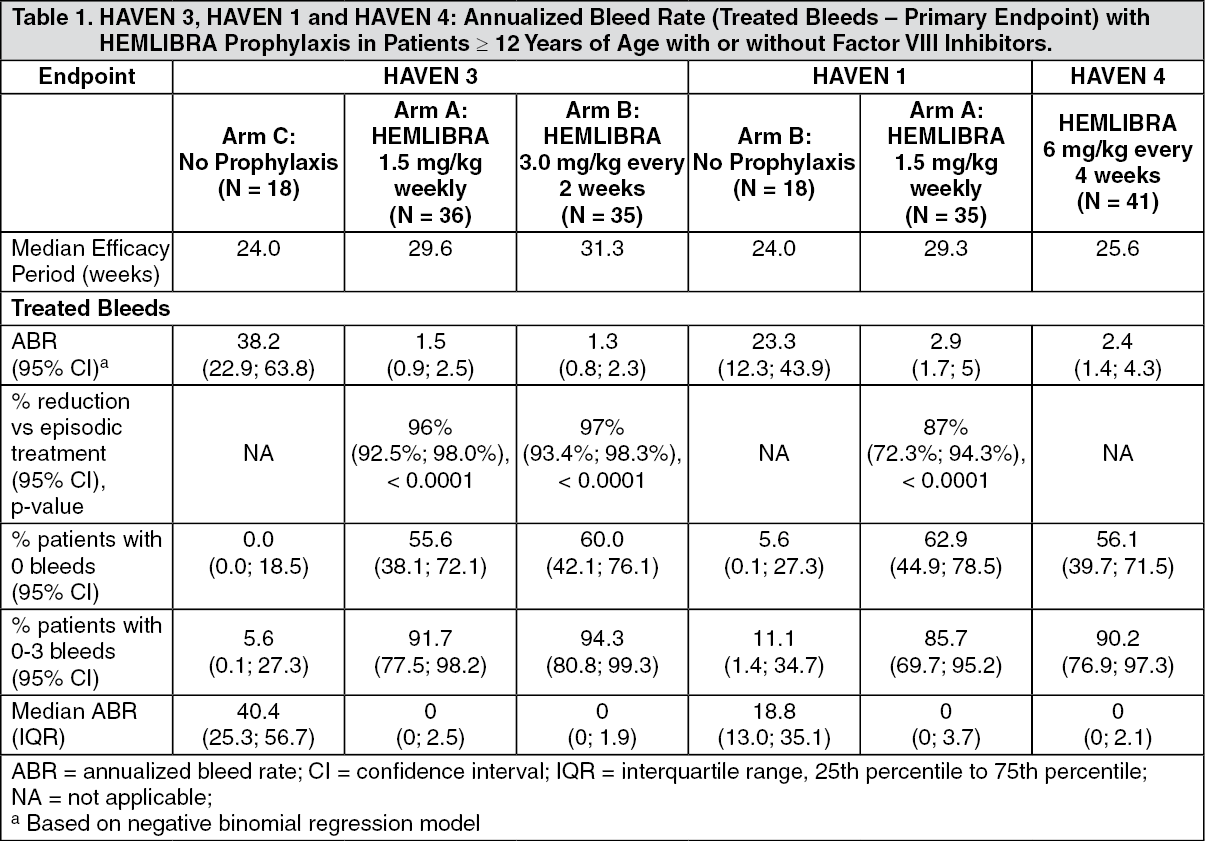
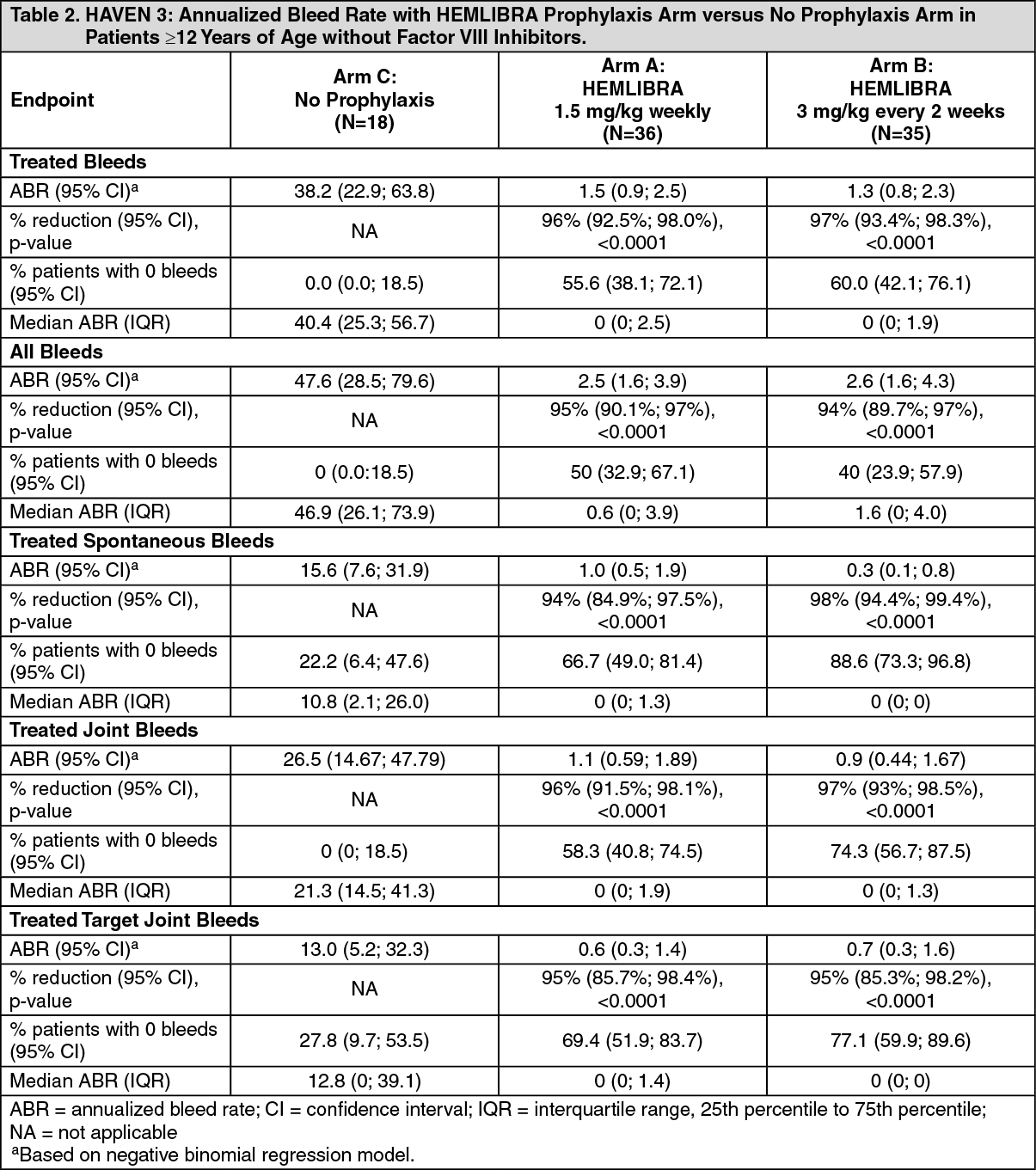
The primary objective of the study was to evaluate in patients previously treated with episodic FVIII the efficacy of prophylactic HEMLIBRA weekly (Arm A) or every two weeks (Arm B) compared to no prophylaxis (Arm C) based on the number of bleeds requiring treatment with coagulation factors (see Table 1). Other objectives of the study included evaluation of the randomized comparison of Arms A or B and Arm C for the efficacy of HEMLIBRA prophylaxis in reducing the number of all bleeds, spontaneous bleeds, joint bleeds, and target joint bleeds (see Table 2). Patient treatment preference was also assessed using a preference survey.
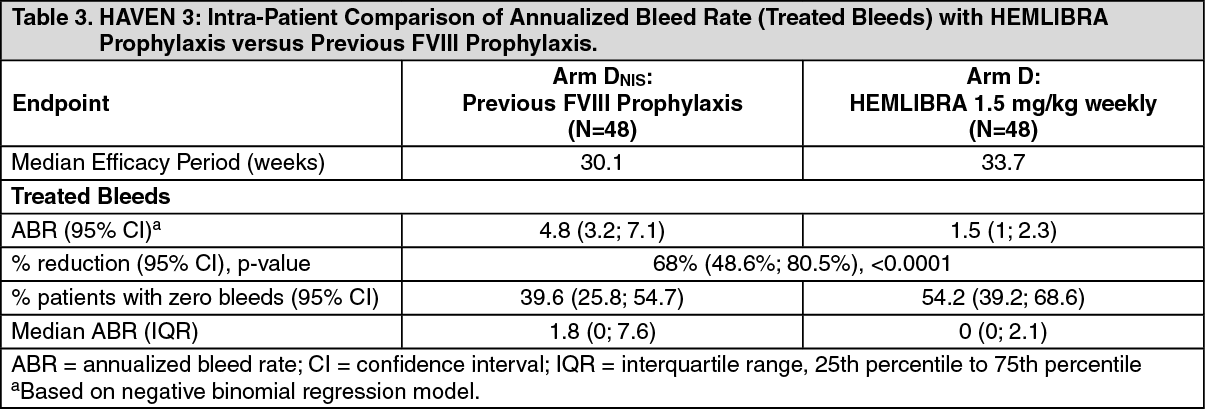
The efficacy of HEMLIBRA prophylaxis was also compared with previous prophylactic FVIII treatment (Arm D) in patients who had participated in a non-interventional study (NIS) prior to enrollment (see Table 3). Only patients from the NIS were included in this comparison, because bleed and treatment data were collected with the same level of granularity as used in HAVEN 3.
HAVEN 1: The HAVEN 1 study was a randomized, multicenter, open-label clinical study in 109 adolescent and adult males (aged ≥ 12 years old and ≥ 40 kg) with hemophilia A with factor VIII inhibitors who had previously received either episodic ("on demand") or prophylactic treatment with bypassing agents. In the study, patients received weekly HEMLIBRA prophylaxis (Arms A, C, and D) - 3 mg/kg once weekly for 4 weeks followed by 1.5 mg/kg once weekly thereafter - or no prophylaxis (Arm B). Patients randomized to Arm B could switch to HEMLIBRA prophylaxis after completing at least 24 weeks without prophylaxis. Dose up-titration to 3 mg/kg once weekly was allowed after 24 weeks on HEMLIBRA prophylaxis for patients who experienced two or more qualified bleeds (i.e., spontaneous and clinically significant bleeds occurring at steady state). During the study, two patients underwent up-titration of their maintenance dose to 3 mg/kg once weekly.
Fifty-three patients previously treated with episodic ("on demand") bypassing agents were randomized in a 2:1 ratio to receive HEMLIBRA prophylaxis (Arm A) or no prophylaxis (Arm B), with stratification by prior 24-week bleed rate (< 9 or ≥ 9).
Forty-nine patients previously treated with prophylactic bypassing agents were enrolled in Arm C to receive HEMLIBRA prophylaxis. Seven patients previously treated with episodic ("on demand") bypassing agents who had participated in the NIS prior to enrollment but were unable to enroll into HAVEN 1 prior to the closure of Arms A and B were enrolled in Arm D to receive HEMLIBRA prophylaxis.
The primary objective of the study was to evaluate among patients previously treated with episodic (on-demand) bypassing agents the treatment effect of weekly HEMLIBRA prophylaxis compared with no prophylaxis (Arm A vs. Arm B) on the number of bleeds requiring treatment with coagulation factors over time (minimum of 24 weeks or date of discontinuation) (see Table 1). Other secondary objectives of the randomized comparison of Arms A and B were the efficacy of weekly HEMLIBRA prophylaxis in reducing the number of all bleeds, spontaneous bleeds, joint bleeds and target joint bleeds (see Table 3), as well as assessing patient-reported health-related quality of life (HRQoL) and health status (see Tables 8 and 9).
The efficacy of weekly HEMLIBRA prophylaxis compared with previous prophylactic bypassing agents was also evaluated in patients who had participated in the NIS prior to enrollment (Arms C) (see Table 5). Only patients from the NIS were included in this comparison, because bleed and treatment data were collected with the same level of granularity as that used in HAVEN 1.
HAVEN 4: HEMLIBRA was investigated in a single arm, multicenter, phase III clinical study in 41 adult and adolescent males (aged ≥ 12 years and ≥ 40 kg) with hemophilia A with or without FVIII inhibitors who previously received either episodic ("on demand") or prophylactic treatment with FVIII or bypassing agents. Patients received HEMLIBRA prophylaxis - 3 mg/kg once weekly for four weeks followed by 6 mg/kg every four weeks thereafter.
The primary objective of the study was to evaluate the efficacy of HEMLIBRA prophylaxis in maintaining adequate bleed control, given every four weeks based on treated bleeds (see Table 1). Other objectives were to evaluate the clinical efficacy of HEMLIBRA prophylaxis on all bleeds, treated spontaneous bleeds, treated joint bleeds and treated target joint bleeds (see Table 7). Patient treatment preference was also assessed using a preference survey.
Adult and Adolescent Efficacy Results: The efficacy results of HEMLIBRA prophylaxis with respect to rate of treated bleeds are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAVEN 3: The efficacy results of HEMLIBRA prophylaxis compared with no prophylaxis with respect to rate of treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are shown as follows in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the HAVEN 3 clinical study intra-patient analysis, HEMLIBRA prophylaxis resulted in a statistically significant (p<0.0001) reduction (68%) in bleed rate for treated bleeds compared with previous FVIII prophylaxis collected in the NIS prior to enrollment (see Table 3).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAVEN 1: The efficacy results of HEMLIBRA prophylaxis compared with no prophylaxis with respect to rate of treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are shown in Table 4. (See Table 4.)
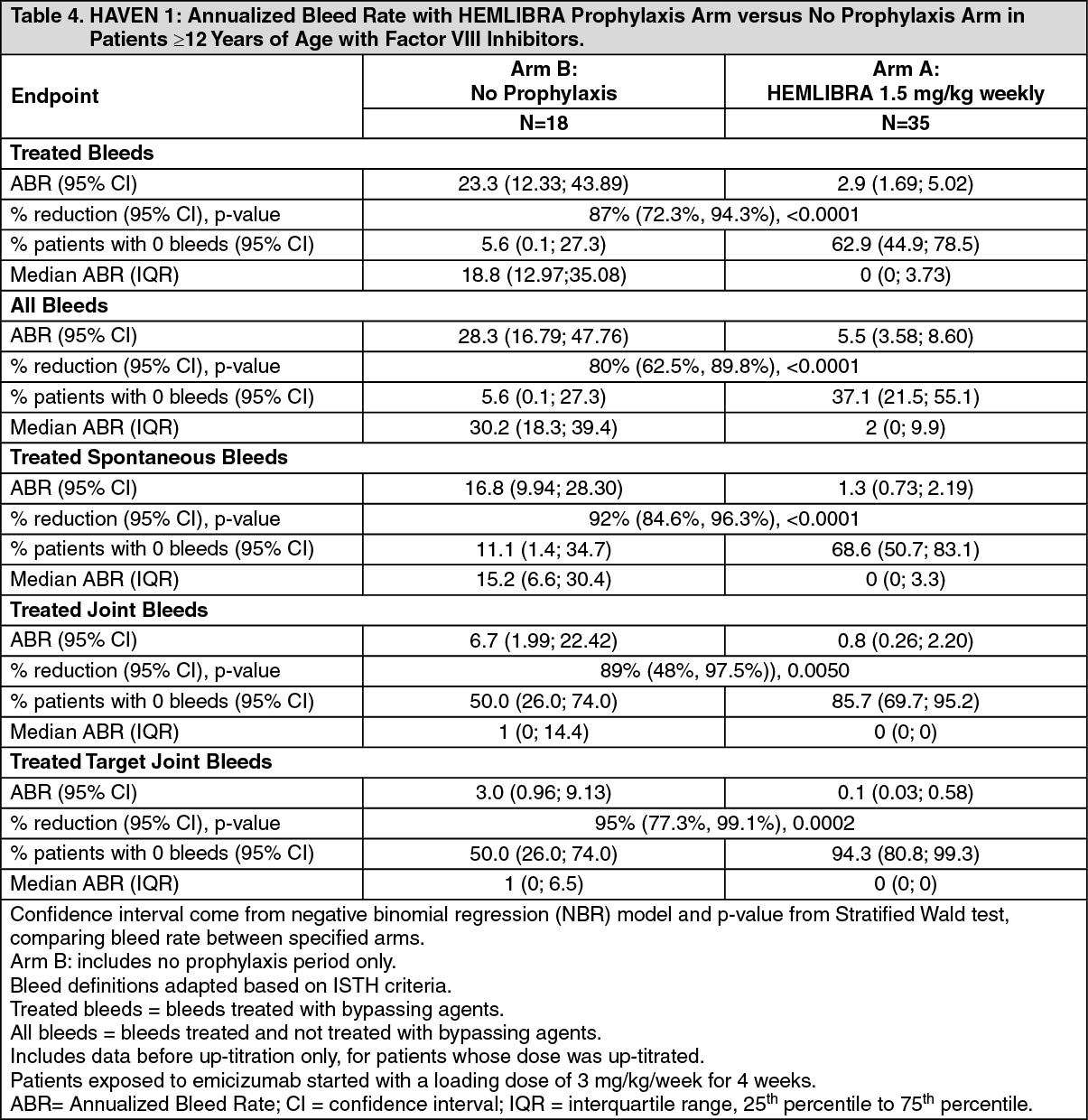 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditional analyses for HAVEN 1 to assess long term control of bleeds with HEMLIBRA prophylaxis were conducted using 12-week treatment intervals up to week 72. When ABR for treated bleeds was assessed over 12-week intervals the mean ABRs decreased over time and the improvement was sustained up to week 72, while the median remained consistently at zero (see Table 5). These data demonstrate the long term efficacy of HEMLIBRA prophylaxis. The mean and median calculated ABRs for treated bleeds are shown in Table 5. (See Table 5.)
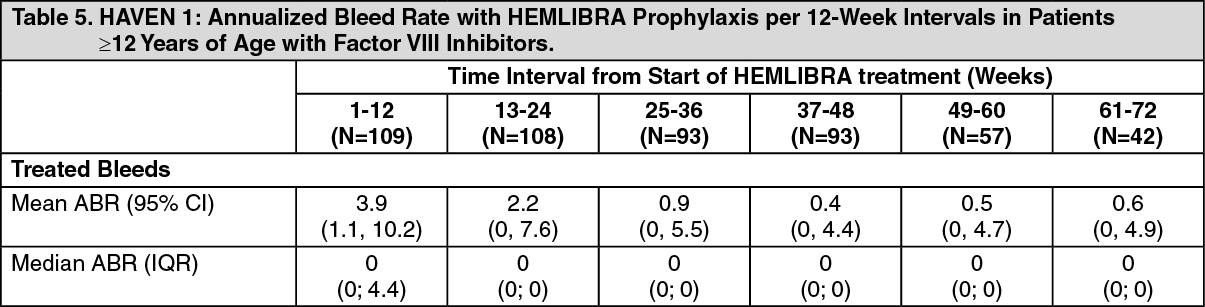 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the HAVEN 1 clinical study intra-patient analysis, HEMLIBRA prophylaxis resulted in a statistically significant (p = 0.0003) reduction (79%) in bleed rate for treated bleeds compared with previous bypassing agent prophylaxis collected in the NIS prior to enrollment (see Table 6).
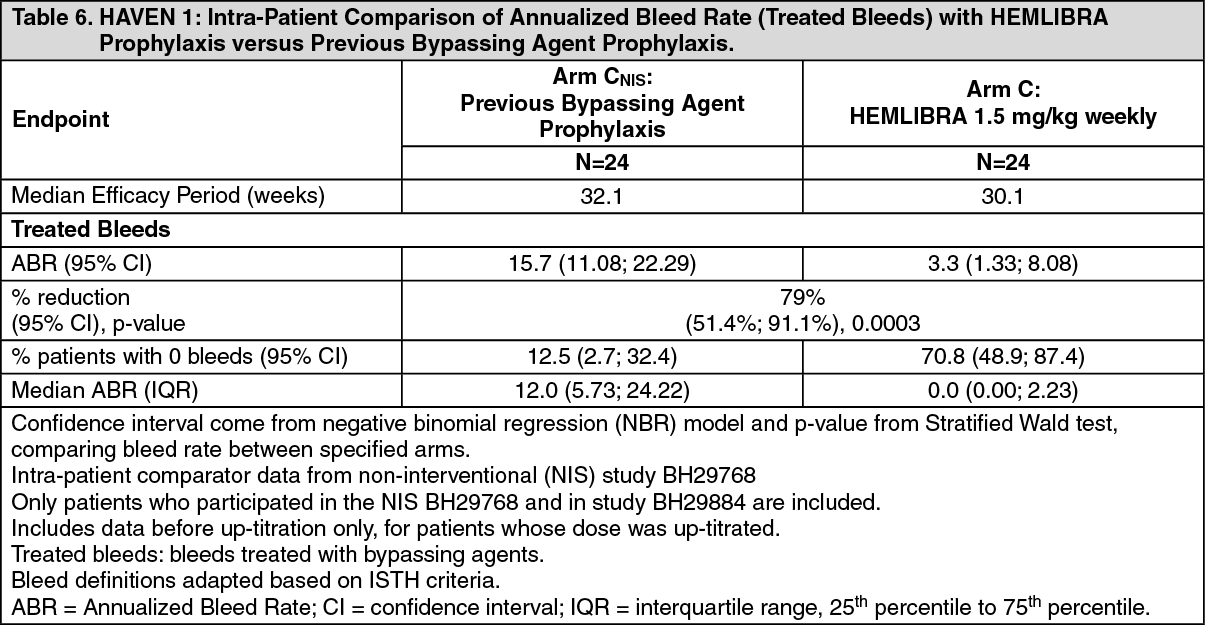 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAVEN 4: Efficacy results for the HAVEN 4 clinical study are summarized as follows. Forty-one patients ≥ 12 years old were evaluated for efficacy with a median observation time of 25.6 weeks (range: 24.1 - 29.4 weeks). The efficacy results of HEMLIBRA prophylaxis every four weeks with respect to rate of treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are shown in Table 7. (See Table 7.)
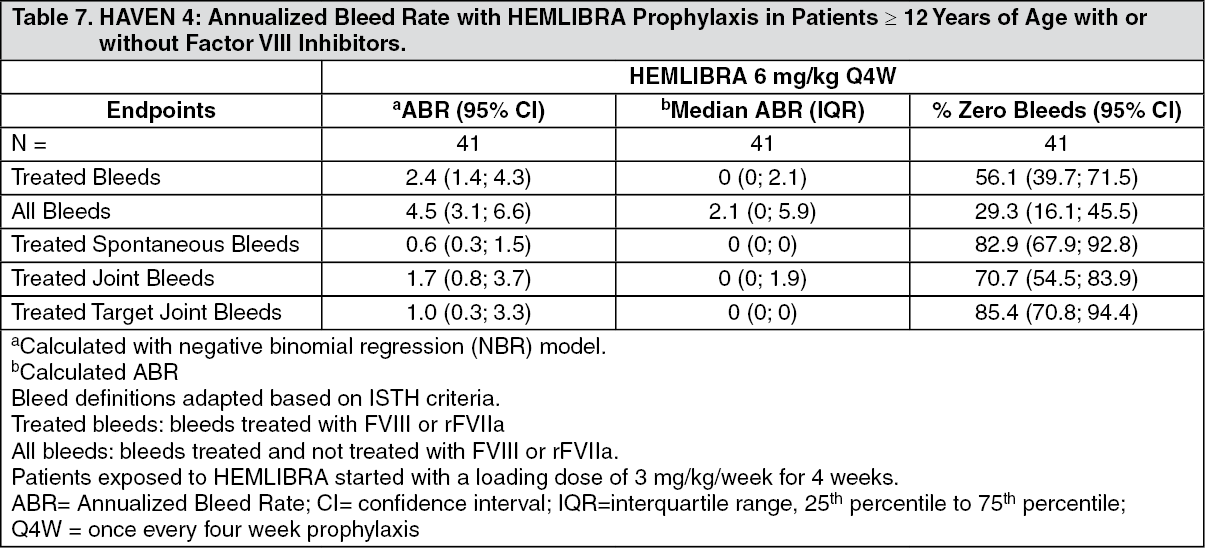 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdults and Adolescents Health-Related Outcome Measures: The HAVEN adult and adolescent clinical studies evaluated patient-reported outcomes with several measures. The Haemophilia-Specific Quality of Life (Haem-A-QoL) questionnaire for adults (≥ 18 years) and its adolescent version (Haemo-QoL-SF, for 8 to <18 years) assessed hemophilia-related quality of life in patients. For the Haem-A-QoL and Haemo-QoL-SF, the Physical Health Score (i.e., painful swellings, presence of joint pain, pain with movement, difficulty walking far and needing more time to get ready) and Total Score (summary of all scores) were protocol defined endpoints of interest. To measure change in health status, the Index Utility Score (IUS) and the Visual Analog Scale (VAS) from the EuroQoL Five-Dimension-Five Levels Questionnaire (EQ-5D-5L) was examined.
In HAVEN 3 and 4, an assessment of patient preference for treatment, the Emicizumab Preference Survey (EmiPref), was used.
Adult and Adolescent Health-Related Outcomes Results: HAVEN 1 Health-Related Outcomes: In HAVEN 1, health-related quality of life (HRQoL) for patients aged ≥ 18 years was evaluated at week 25 based on the Haem-A-QoL questionnaire for adults (see Table 8). The Haem-A-QoL is a valid and reliable measure of HRQoL. (See Table 8.)
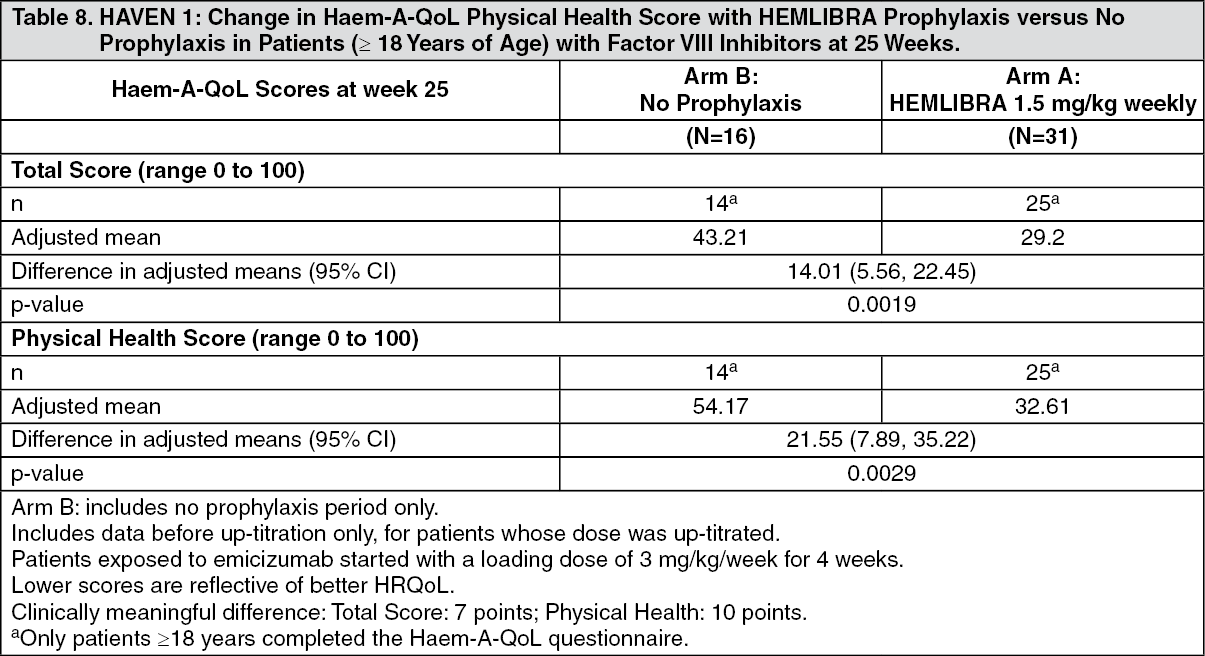 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAVEN 1 Health Status Outcomes: In HAVEN 1, patients' health status was assessed according to EQ-5D-5L. EQ-5D-5L is a valid and reliable measure of health status (see Table 9).
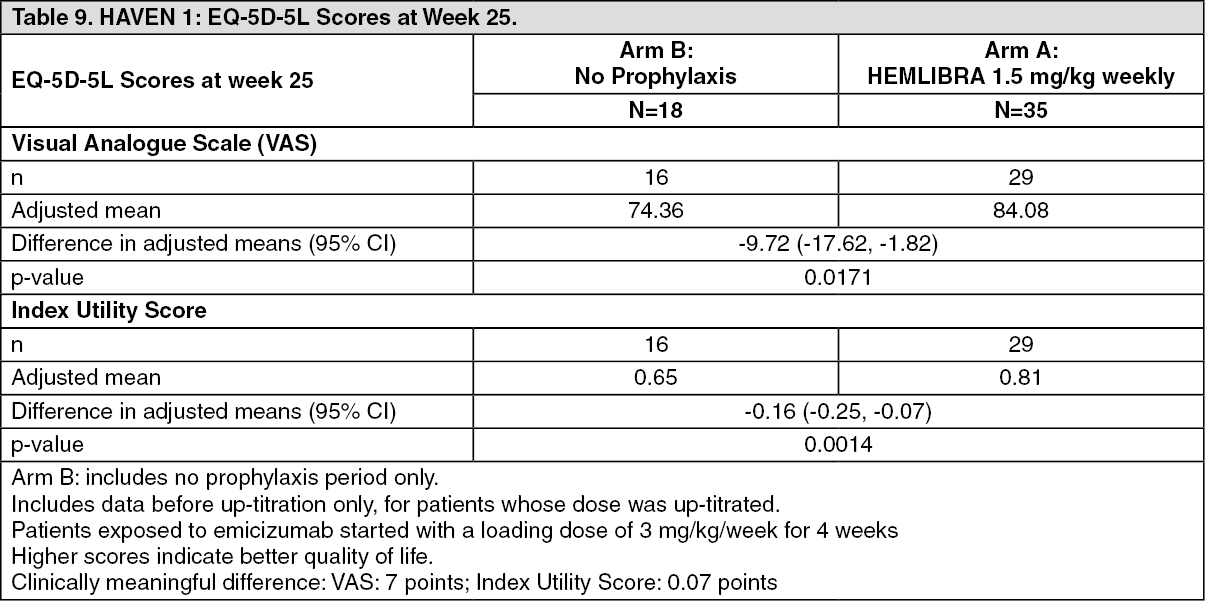 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHAVEN 3 and 4 Patient Preference: In HAVEN 3 and HAVEN 4, patients who received HEMLIBRA (once weekly, every two weeks or every four weeks) reported whether they preferred subcutaneous HEMLIBRA, their prior IV treatment or had no preference at week 17. Of the patients in HAVEN 3 who responded to the preference questionnaire, 89 of 95 patients (93.7%) reported preferring HEMLIBRA to their prior IV treatment, and specifically 45 of 46 patients (97.8%) preferred HEMLIBRA to their prior prophylactic FVIII treatment. In HAVEN 4, all 41 patients (100%) responded to the preference questionnaire and reported preferring HEMLIBRA to their prior IV treatment.
In HAVEN 3 and 4, the two reasons most frequently ranked by patients as the most important for their preference for HEMLIBRA were that the route of administration was easier and the frequency of treatments was lower.
Clinical Study in Pediatric Patients: HAVEN 2 (Interim Analysis): HEMLIBRA weekly prophylaxis was evaluated in a single-arm, multicenter, open-label clinical study in pediatric patients (age < 12 years old, or 12 to 17 years old weighing < 40 kg) with hemophilia A with factor VIII inhibitors. Patients received HEMLIBRA prophylaxis at 3 mg/kg once weekly for the first four weeks followed by 1.5 mg/kg once weekly thereafter.
The study evaluated the pharmacokinetics, safety, and efficacy including the efficacy of weekly HEMLIBRA prophylaxis compared with previous episodic and prophylactic bypassing agent treatment in patients who had participated in the NIS prior to enrollment (intra-patient comparison).
HAVEN 2 Efficacy Results (Interim Analysis): At the time of the interim analysis, efficacy was evaluated in 59 pediatric patients who were < 12 years of age and had been receiving weekly HEMLIBRA prophylaxis for at least 12 weeks, including 38 patients age 6 to < 12 years; 17 patients age 2 to < 6 years and four patients < 2 years old. Annualized bleed rate and percent of patients with zero bleeds were calculated for 59 patients (see Table 10). The median observation time for these patients was 29.6 weeks (range: 18.4 - 63). (See Table 10.)
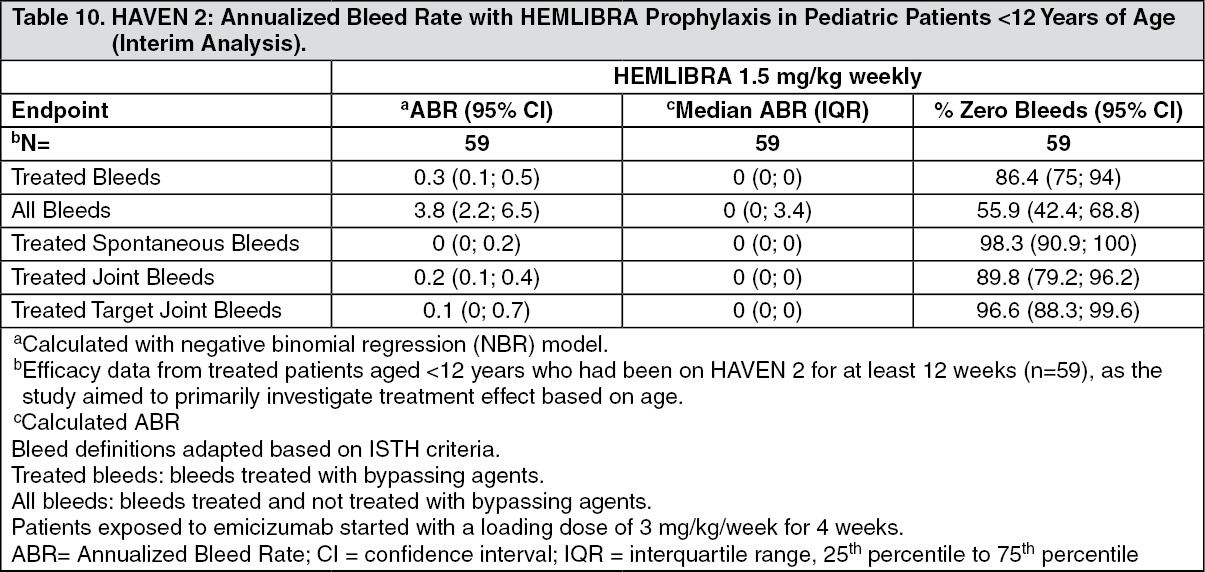 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the intra-patient interim analysis, weekly HEMLIBRA prophylaxis resulted in a clinically meaningful (98%) reduction in bleed rate for treated bleeds in 18 pediatric patients who had at least 12 weeks of HEMLIBRA prophylaxis compared to their bleed rate collected in the NIS prior to enrollment (see Table 11).
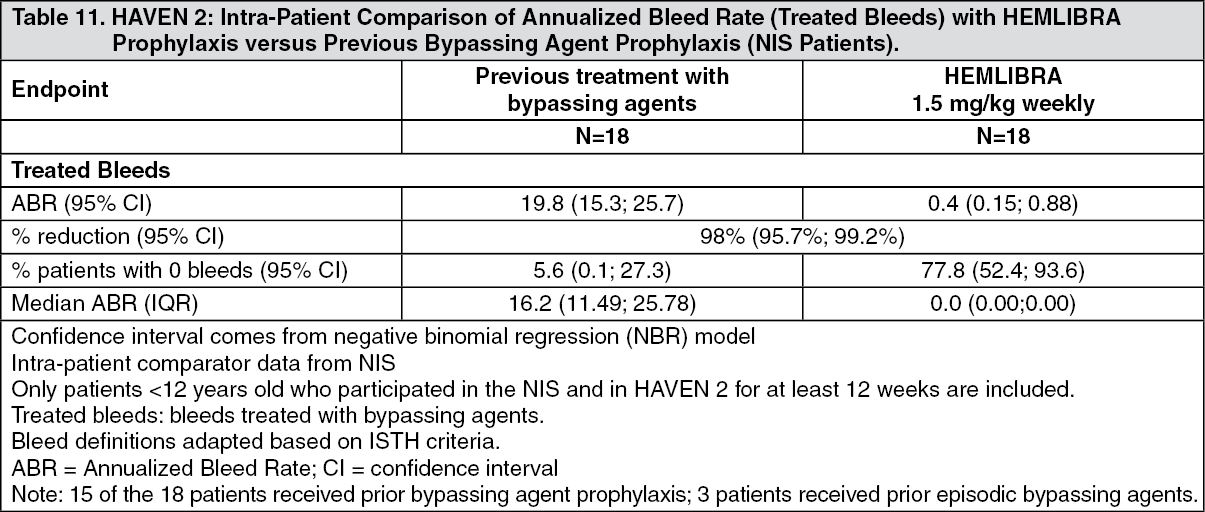 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePediatric Health-Related Outcomes Results: HAVEN 2 Health-Related Outcomes: In HAVEN 2, HRQoL for patients aged ≥ 8 to < 12 years was evaluated at week 25 based on the Haemo-QoL-SF questionnaire for children. The Haemo-QoL-SF is a valid and reliable measure of HRQoL (see Table 12).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn HAVEN 2, HRQoL for patients ages < 12 years was also evaluated at week 25 based on the Adapted InhibQoL with Aspects of Caregiver Burden questionnaire completed by caregivers. The Adapted InhibQoL is a valid and reliable measure of HRQoL (see Table 13).
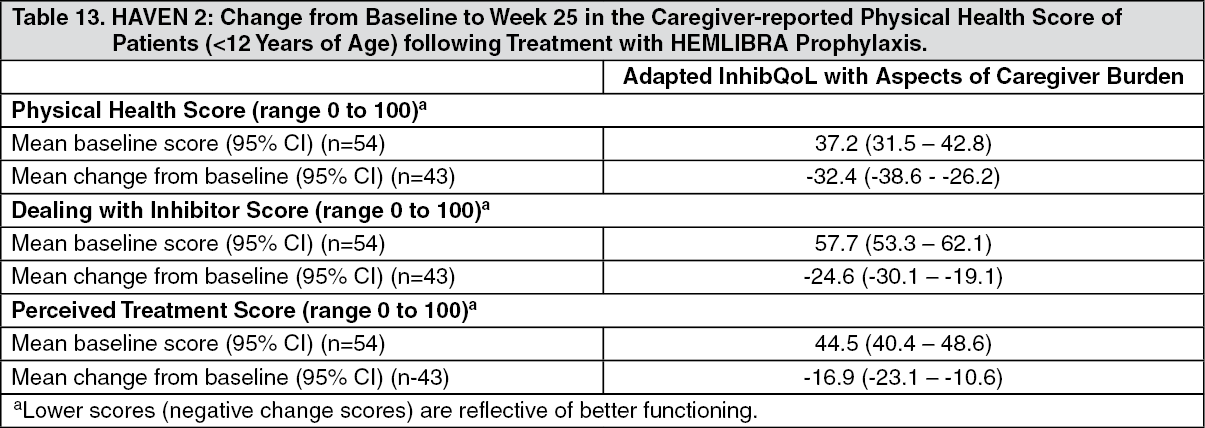 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSurgeries and Procedures in the HAVEN Clinical Studies: There is limited experience on bypassing agent or FVIII use during surgeries and procedures in patient receiving HEMLIBRA prophylaxis. In the clinical studies, bypassing agent or FVIII use during surgeries and procedures was determined by the investigator.
IMMUNOGENICITY: As with all therapeutic proteins, there is the potential for an immune response in patients treated with HEMLIBRA. A total of 668 patients were tested for anti-emicizumab antibodies in the pooled phase III clinical trials, of which 34 patients (5.1%) tested positive for anti-emicizumab antibodies. In 18 patients (2.7%), antiemicizumab antibodies were neutralizing in vitro. Of these, the neutralizing antiemicizumab antibodies did not appear to have a clinically meaningful impact on the pharmacokinetics or efficacy of HEMLIBRA in 14 patients, while decreased emicizumab plasma concentrations were observed in four patients (0.6%). One patient (0.2%) with neutralizing anti-emicizumab antibodies and decreased emicizumab plasma concentrations, experienced loss of efficacy after 5 weeks of treatment and discontinued HEMLIBRA. Overall, the safety profile of HEMLIBRA was similar between those patients with anti-emicizumab antibodies (including neutralizing antibodies) and those without (see Precautions and CLINICAL TRIALS under Adverse Reactions).
The data reflect the number of patients whose test results were considered positive for antibodies to emicizumab using an enzyme-linked immunosorbent assay (ELISA) and/or for neutralizing anti-emicizumab antibodies using a FVIII chromogenic assay. Immunogenicity assay results may be influenced by several factors including assay sensitivity and specificity, sample handling, timing of sample collection, concomitant medicinal products and underlying disease. For these reasons, comparison of incidence of antibodies to emicizumab with the incidence of antibodies to other products may be misleading.
PHARMACOKINETICS: The pharmacokinetics of emicizumab were determined via a non-compartmental analysis in healthy subjects and using a population pharmacokinetic analysis on a database composed of 389 patients with hemophilia A.
ABSORPTION: Following subcutaneous administration in hemophilia A patients, the absorption half-life was 1.6 days.
Following multiple subcutaneous administrations of 3 mg/kg once weekly for the first 4 weeks in hemophilia A patients, mean (±SD) trough plasma concentrations of emicizumab achieved 52.6±13.6 μg/mL at week 5. Sustained mean trough plasma concentrations of emicizumab at steady-state were 51.2 μg/mL, 46.9 μg/mL and 38.5 μg/mL with the recommended maintenance doses of 1.5 mg/kg once weekly, 3 mg/kg every two weeks or 6 mg/kg every four weeks, respectively (Figure 1, Table 14). (See Figure 1.)
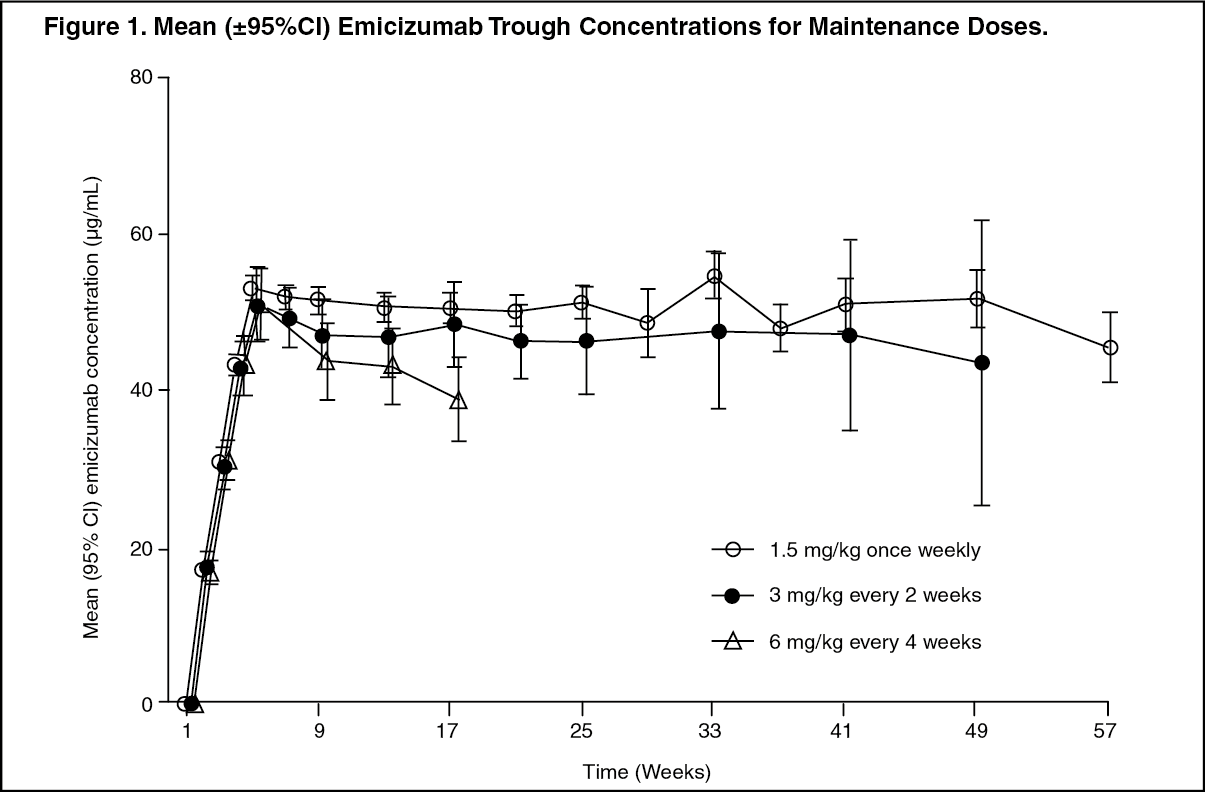 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe mean (±SD) Ctrough, Cmax and ratios of Cmax/Ctrough at steady-state for the recommended maintenance doses of 1.5 mg/kg once weekly, 3 mg/kg every two weeks or 6 mg/kg every four weeks are shown in Table 14. (See Table 14.)
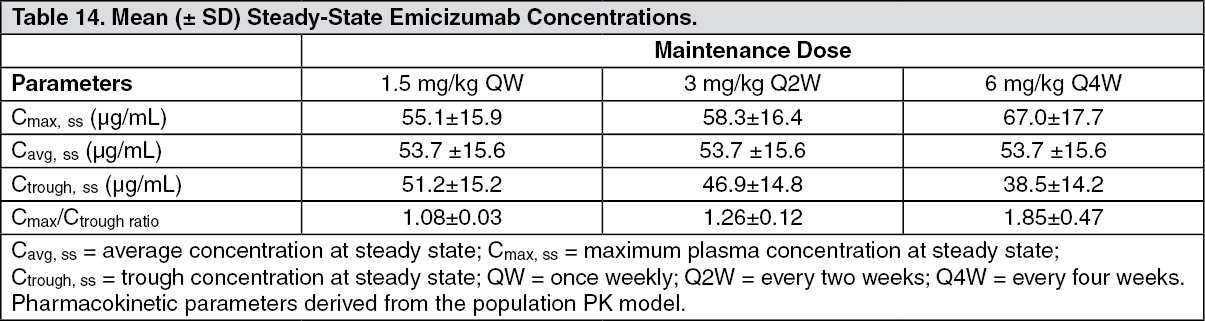 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSimilar PK profiles were observed following once weekly dosing (3 mg/kg/week for 4 weeks followed by 1.5 mg/kg/week) in adults/adolescents (≥ 12 years) and children (< 12 years) (see Figure 2).
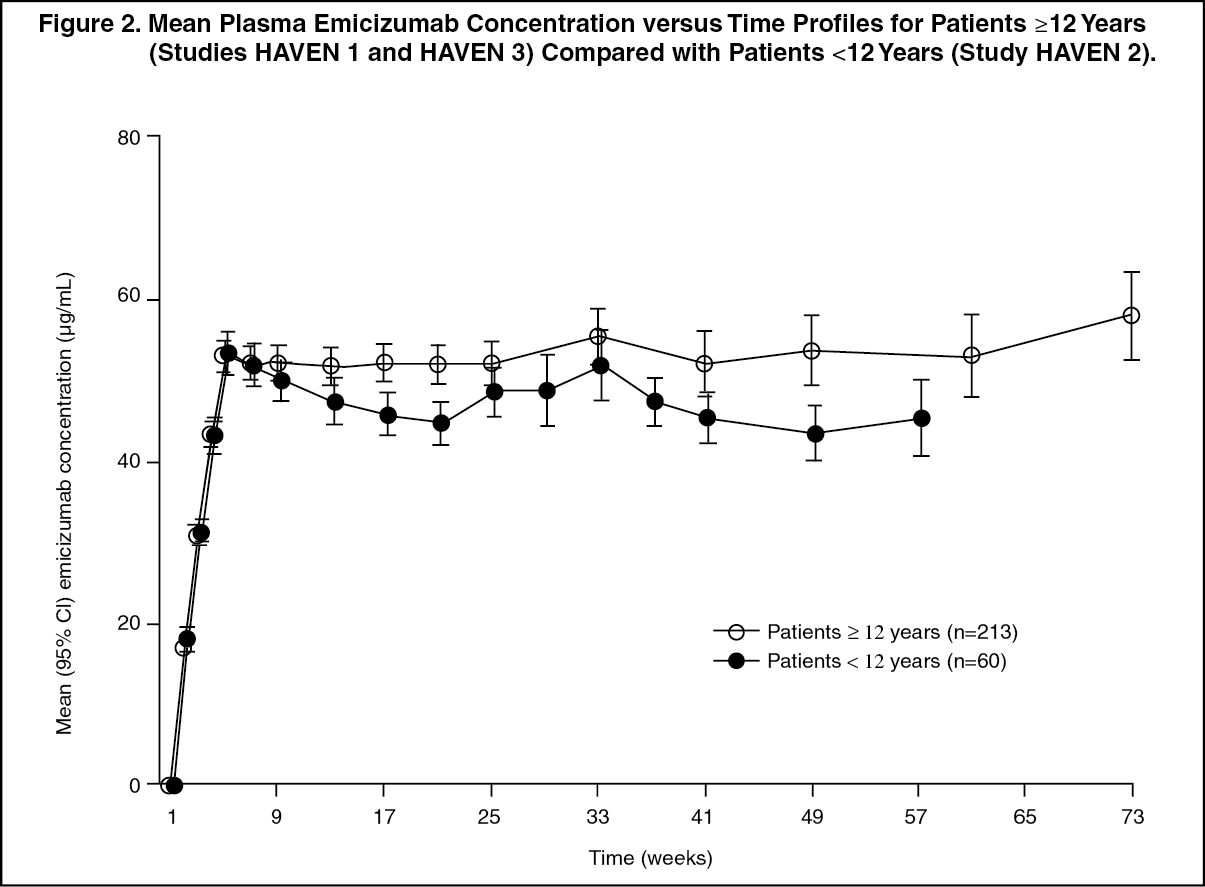 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn healthy subjects, the absolute bioavailability following subcutaneous administration of 1 mg/kg was between 80.4% and 93.1% depending on the injection site. Similar pharmacokinetic profiles were observed following subcutaneous administration in the abdomen, upper arm, and thigh. Emicizumab can be administered interchangeably at these anatomical sites (see Dosage & Administration).
DISTRIBUTION: Following a single intravenous dose of 0.25 mg/kg emicizumab in healthy subjects, the volume of distribution at steady state was 106 mL/kg (i.e., 7.4 L for a 70-kg adult). Emicizumab is not intended for intravenous use (see Dosage & Administration).
The apparent volume of distribution (V/F), estimated from the population PK analysis, in hemophilia A patients following multiple subcutaneous doses of emicizumab was 10.4 L.
METABOLISM: The metabolism of emicizumab has not been studied. IgG antibodies are mainly catabolized by lysosomal proteolysis and then eliminated from or reused by the body.
ELIMINATION: Following intravenous administration of 0.25 mg/kg in healthy subjects, the total clearance of emicizumab was 3.26 mL/kg/day (i.e. 0.228 L/d for a 70-kg adult) and the mean terminal half-life was 26.7 days.
Following single subcutaneous injection in healthy subjects, the elimination half-life was approximately 4 to 5 weeks.
Following multiple subcutaneous injections in hemophilia A patients, the apparent clearance was 0.271 L/day and the elimination apparent half-life was 26.9 days.
Dose Linearity: Emicizumab exhibited dose-proportional pharmacokinetics in patients with hemophilia A over a dose range from 0.3 to 6 mg/kg once weekly following subcutaneous administration.
PHARMACOKINETICS IN SPECIAL POPULATIONS: Renal impairment: No dedicated studies on the effect of renal impairment on the pharmacokinetics of emicizumab have been conducted. Most of the patients with hemophilia A in the population pharmacokinetic analysis had normal renal function (N = 332; creatinine clearance [CLcr] ≥ 90 mL/min) or mild renal impairment (N = 27; CLcr of 60-89 mL/min). Only 2 patients had moderate renal impairment (CLcr of 30-59 mL/min). No patients had severe renal impairment. Mild or moderate renal impairment did not appear to have an impact on the pharmacokinetics of emicizumab (see also SPECIAL DOSAGE INSTRUCTIONS under Dosage & Administration).
Hepatic impairment: No dedicated studies on the effect of hepatic impairment on the pharmacokinetics of emicizumab have been conducted. Most of the patients with hemophilia A in the population pharmacokinetic analysis had normal hepatic function (bilirubin and AST ≤ ULN, N = 300) or mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin from 1.0 to 1.5 x ULN and any AST, N = 51). Only 6 patients had moderate hepatic impairment (1.5 x ULN < bilirubin ≤ 3 x ULN and any AST). Mild or moderate hepatic impairment did not affect the pharmacokinetics of emicizumab (see also SPECIAL DOSAGE INSTRUCTIONS under Dosage & Administration). Hepatic impairment was defined by the National Cancer Institute (NCI) criteria for hepatic dysfunction.
Pediatrics: The effect of age on the pharmacokinetics of emicizumab was assessed in a population pharmacokinetic analysis which included 5 infants (≥ 1 month to < 2 years), 55 children (≥ 2 years to < 12 years) and 50 adolescents (12 to < 18 years) with hemophilia A. Age did not affect the pharmacokinetics of emicizumab in pediatric patients (see SPECIAL DOSAGE INSTRUCTIONS under Dosage & Administration).
Geriatrics: The effect of age on the pharmacokinetics of emicizumab was assessed in a population pharmacokinetic analysis which included 13 patients aged 65 years and older (no patients were older than 77 years of age). Relative bioavailability decreased with older age, but no clinically important differences were observed in the pharmacokinetics of emicizumab between patients < 65 years and patients ≥ 65 years.
Race: Population pharmacokinetics analyses in patients with hemophilia A showed that race did not affect the pharmacokinetics of emicizumab.
Toxicology: PRECLINICAL SAFETY: Preclinical data reveal no special hazards for humans based on studies of acute and repeated dose toxicity, including safety pharmacology endpoints and endpoints for reproductive toxicity.
CARCINOGENICITY: No carcinogenicity studies have been performed to establish the carcinogenic potential of emicizumab.
GENOTOXICITY: No studies have been performed to establish the mutagenic potential of emicizumab.
IMPAIRMENT OF FERTILITY: Emicizumab did not cause any toxicological changes in the reproductive organs of male or female cynomolgus monkeys at doses of up to 30 mg/kg/week in subcutaneous general toxicity studies of up to 26-week duration and at doses of up to 100 mg/kg/week in a 4-week intravenous general toxicity study.
REPRODUCTIVE TOXICITY: No data are available with respect to potential side effects of emicizumab on embryo-fetal development.
OTHER: In an in vitro study of cytokine release that used the whole blood of healthy adults, the levels of cytokines induced by emicizumab were comparable to those induced by other low-risk antibodies.