


 Sign Out
Sign Out
Advise patients/caregivers to record the batch number of the product whenever HEMLIBRA is administered outside of a healthcare setting.
Thrombotic microangiopathy associated with HEMLIBRA and activated prothrombin complex concentrate: Cases of thrombotic microangiopathy (TMA) were reported from a clinical trial in patients receiving HEMLIBRA prophylaxis when on average a cumulative amount of > 100 U/kg/24 hours of activated prothrombin complex concentrate (aPCC) for 24 hours or more were administered (see CLINICAL TRIALS under Adverse Reactions). Treatment for the TMA events included supportive care with or without plasmapheresis and hemodialysis. Evidence of improvement was seen within one week following discontinuation of aPCC. This rapid clinical improvement is distinct from the usual clinical course observed in atypical hemolytic uremic syndrome and classic TMAs such as thrombotic thrombocytopenic purpura (see CLINICAL TRIALS under Adverse Reactions).
Patients receiving HEMLIBRA prophylaxis should be monitored for the development of TMA when administering aPCC. The physician should immediately discontinue aPCC and interrupt HEMLIBRA therapy if clinical symptoms and/or laboratory findings consistent with TMA occur, and manage as clinically indicated. Physicians and patients/caregivers should weigh the benefits and risks of resuming HEMLIBRA prophylaxis following complete resolution of TMA on a case-by-case basis. In case a bypassing agent is indicated in a patient receiving HEMLIBRA prophylaxis, see subsection as follows for dosing recommendations for the use of bypassing agents.
Thromboembolism associated with HEMLIBRA and activated prothrombin complex concentrate: Thrombotic events were reported from a clinical trial in patients receiving HEMLIBRA prophylaxis when on average a cumulative amount of > 100 U/kg/24 hours of aPCC for 24 hours or more were administered (see CLINICAL TRIALS under Adverse Reactions). No cases required anticoagulation therapy, which is distinct from the usual treatment of thrombotic events. Evidence of improvement or resolution was seen after discontinuation of aPCC (see CLINICAL TRIALS under Adverse Reactions).
Patients receiving HEMLIBRA prophylaxis should be monitored for the development of thromboembolism when administering aPCC. The physician should immediately discontinue aPCC and interrupt HEMLIBRA therapy if clinical symptoms, imaging, and/or laboratory findings consistent with thrombotic events occur, and manage as clinically indicated. Physicians and patients/caregivers should weigh the benefits and risks of resuming HEMLIBRA prophylaxis following complete resolution of thrombotic events on a case-by-case basis. In case a bypassing agent is indicated in a patient receiving HEMLIBRA prophylaxis, see subsection as follows for dosing recommendations for the use of bypassing agents.
Guidance on the use of bypassing agents in patients receiving HEMLIBRA prophylaxis: Treatment with bypassing agents should be discontinued the day before starting HEMLIBRA therapy.
Physicians should discuss with all patients and/or caregivers the exact dose and schedule of bypassing agents to use, if required, while receiving HEMLIBRA prophylaxis.
HEMLIBRA increases the patient's coagulation potential. The bypassing agent dose required may therefore be lower than that used without HEMLIBRA prophylaxis. The dose and duration of treatment with bypassing agents will depend on the location and extent of bleeding and on the patient's clinical condition. Avoid use of aPCC unless no other treatment options/alternatives are available. If aPCC is indicated in a patient receiving HEMLIBRA prophylaxis the initial dose should not exceed 50 U/kg. If bleeding is not controlled with the initial dose of aPCC up to 50 U/kg, additional aPCC doses should be administered under medical guidance or supervision, and the total aPCC dose should not exceed 100 U/kg in the first 24-hours of treatment. Treating physicians must carefully weigh the risk of TMA and thromboembolism against the risk of bleeding when considering aPCC treatment beyond a maximum of 100 U/kg in the first 24-hours.
In clinical trials, no cases of TMA or thrombotic events were observed with use of activated recombinant human FVII (rFVIIa) alone in patients receiving HEMLIBRA prophylaxis.
Bypassing agent dosing guidance should be followed for at least 6 months following discontinuation of HEMLIBRA prophylaxis (see PHARMACOLOGY: PHARMACOKINETICS: ELIMINATION under Actions).
Immunogenicity: Anti-emicizumab antibodies have been reported in a small number of patients treated with HEMLIBRA in clinical trials. Most patients found to have anti-emicizumab antibodies did not experience a change in emicizumab plasma concentrations or an increase in bleeding events; however, in uncommon (≥ 1/1,000 to < 1/100) cases, the presence of neutralizing anti-emicizumab antibodies with decreasing emicizumab concentration may be associated with loss of efficacy (see CLINICAL TRIALS under Adverse Reactions and PHARMACOLOGY: PHARMACODYNAMICS: IMMUNOGENICITY under Actions).
In case of clinical signs of loss of efficacy (e.g. increase in breakthrough bleeding events), prompt evaluation by a physician should be sought to assess the etiology and a possible change in treatment should be considered.
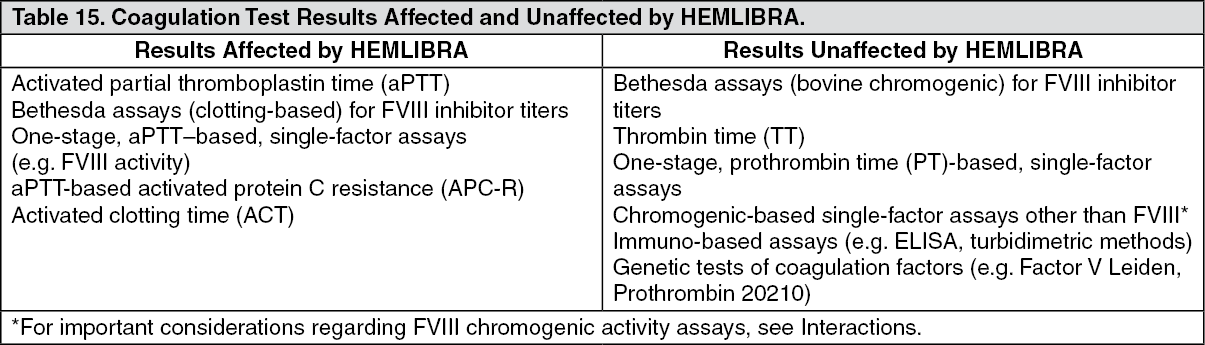
Laboratory coagulation test interference: HEMLIBRA affects intrinsic pathway clotting-based laboratory tests, including the activated clotting time (ACT), activated partial thromboplastin time (aPTT) and all assays based on aPTT, such as one-stage factor VIII activity (see Table 15 as follows). Therefore, intrinsic pathway clotting-based laboratory test results in patients treated with HEMLIBRA should not be used to monitor HEMLIBRA activity, determine dosing for factor replacement or anti-coagulation, or measure factor VIII inhibitor titers. Laboratory tests affected and unaffected by HEMLIBRA are shown in Table 15 as follows (see Interactions). (See Table 15.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDRUG ABUSE AND DEPENDENCE: HEMLIBRA does not have the potential for abuse and dependence.
ABILITY TO DRIVE AND USE MACHINES: There is no evidence that treatment with HEMLIBRA results in an increase in adverse reactions that might lead to the impairment of the ability to drive and use machines.
RENAL IMPAIRMENT: The safety and efficacy of HEMLIBRA have not been specifically tested in patients with renal impairment. There are limited data available on the use of HEMLIBRA in patients with mild to moderate renal impairment. No data are available on the use of HEMLIBRA in patients with severe renal impairment. HEMLIBRA is a monoclonal antibody and is cleared via catabolism rather than by renal excretion and a change in dose is not expected to be required for patients with renal impairment (see SPECIAL DOSAGE INSTRUCTIONS under Dosage & Administration and PHARMACOLOGY: PHARMACOKINETICS: PHARMACOKINETICS IN SPECIAL POPULATIONS under Actions).
HEPATIC IMPAIRMENT: The safety and efficacy of HEMLIBRA have not been specifically tested in patients with hepatic impairment. Patients with mild and moderate hepatic impairment were included in clinical trials. No data are available on the use of HEMLIBRA in patients with severe hepatic impairment. HEMLIBRA is a monoclonal antibody and is cleared via catabolism rather than by hepatic metabolism and a change in dose is not expected to be required for patients with hepatic impairment (see SPECIAL DOSAGE INSTRUCTIONS under Dosage & Administration and PHARMACOLOGY: PHARMACOKINETICS: PHARMACOKINETICS IN SPECIAL POPULATIONS under Actions).
USE IN CHILDREN: The safety and efficacy of HEMLIBRA have been established in pediatric patients. Use of HEMLIBRA in pediatric patients with hemophilia A (with or without FVIII inhibitors) is supported by two randomized studies (HAVEN 3 and HAVEN 1) and two single-arm studies (HAVEN 4 and HAVEN 2).
These four clinical studies included a total of 107 pediatric patients in the following age groups: 47 adolescents (12 years to < 18 years), 55 children (2 years to < 12 years) and 5 infants (1 month to < 2 years) (see PHARMACOLOGY: PHARMACODYNAMICS: CLINICAL/EFFICACY STUDIES under Actions). Safety and efficacy results were consistent with those observed for adults (see SPECIAL DOSAGE INSTRUCTIONS under Dosage & Administration, PHARMACOLOGY: PHARMACOKINETICS: PHARMACOKINETICS IN SPECIAL POPULATIONS under Actions and CLINICAL TRIALS under Adverse Reactions).
The steady-state plasma trough concentrations of emicizumab were comparable in adult and pediatric patients at equivalent weight-based doses (PHARMACOLOGY: PHARMACOKINETICS: PHARMACOKINETICS IN SPECIAL POPULATIONS under Actions).
USE IN THE ELDERLY: The safety and efficacy of HEMLIBRA have not been specifically tested in a geriatric population. Clinical studies of HEMLIBRA included 13 patients aged 65 and over. Relative bioavailability decreased with older age, but no clinically important differences were observed in the pharmacokinetics of emicizumab between patients < 65 years and patients ≥ 65 years (see PHARMACOLOGY: PHARMACOKINETICS: PHARMACOKINETICS IN SPECIAL POPULATIONS under Actions).