


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Ribociclib is a selective inhibitor of cyclin-dependent kinase (CDK) 4 and 6, resulting in 50% inhibition (IC50) values of 0.01 (4.3 ng/ml) and 0.039 μM (16.9 ng/ml) in biochemical assays, respectively. These kinases are activated upon binding to D-cyclins and play a crucial role in signalling pathways which lead to cell cycle progression and cellular proliferation. The cyclin D-CDK4/6 complex regulates cell cycle progression through phosphorylation of the retinoblastoma protein (pRb).
In vitro, ribociclib decreased pRb phosphorylation, leading to arrest in the G1 phase of the cell cycle, and reduced cell proliferation in breast cancer cell lines. In vivo, treatment with single-agent ribociclib led to tumour regressions which correlated with inhibition of pRb phosphorylation.
In vivo studies using patient-derived oestrogen receptor-positive breast cancer xenograft model combinations of ribociclib and antioestrogens (i.e. letrozole) resulted in superior tumour growth inhibition with sustained tumour regression and delayed tumour regrowth after stopping dosing compared to each substance alone. Additionally, in vivo antitumour activity of ribociclib in combination with fulvestrant was assessed in immune-deficient mice bearing the ZR751 ER+ human breast cancer xenografts and the combination with fulvestrant resulted in complete tumour growth inhibition.
When tested in a panel of breast cancer cell lines with known ER status, ribociclib demonstrated to be more efficacious in ER+ breast cancer cell lines than in the ER- ones. In the preclinical models tested so far, intact pRb was required for ribociclib activity.
Cardiac electrophysiology: Serial, triplicate ECGs were collected following a single dose and at steady state to evaluate the effect of ribociclib on the QTc interval in patients with advanced cancer. A pharmacokinetic-pharmacodynamic analysis included a total of 997 patients treated with ribociclib at doses ranging from 50 to 1200 mg. The analysis suggested that ribociclib causes concentration-dependent increases in the QTc interval. The estimated QTcF mean change from baseline for 600 mg Kisqali in combination with NSAI or fulvestrant was 22.0 msec (90% CI: 20.56, 23.44) and 23.7 msec (90% CI: 22.31, 25.08), respectively at the geometric mean Cmax at steady-state compared to 34.7 msec (90% CI: 31.64, 37.78) in combination with tamoxifen (see Precautions).
Clinical efficacy and safety: Study CLEE011A2301 (MONALEESA-2): Kisqali was evaluated in a randomised, double-blind, placebo-controlled, multicentre phase III clinical study in the treatment of postmenopausal women with hormone receptor-positive, HER2-negative, advanced breast cancer who received no prior therapy for advanced disease in combination with letrozole versus letrozole alone.
A total of 668 patients were randomised in a 1:1 ratio to receive either Kisqali 600 mg and letrozole (n=334) or placebo and letrozole (n=334), stratified according to the presence of liver and/or lung metastases (Yes [n=292 (44%)]) versus No [n=376 (56%)]). Demographics and baseline disease characteristics were balanced and comparable between study arms. Kisqali was given orally at a dose of 600 mg daily for 21 consecutive days followed by 7 days off treatment in combination with letrozole 2.5 mg once daily for 28 days. Patients were not allowed to cross over from placebo to Kisqali during the study or after progression of disease.
Patients enrolled in this study had a median age of 62 years (range 23 to 91). 44.2% patients were older than 65 years, including 69 patients older than 75 years. The patients included were Caucasian (82.2%), Asian (7.6%), and Black (2.5%). All patients had an ECOG performance status of 0 or 1. In the Kisqali arm 43.7% of patients had received chemotherapy in the neoadjuvant or adjuvant setting and 52.4% had received antihormonal therapy in the neoadjuvant or adjuvant setting prior to study entry. 34.1% of patients were de novo. 20.7% of patients had bone-only disease and 59.0% of patients had visceral disease. Patients with prior (neo)adjuvant therapy with anastrozole or letrozole must have completed this therapy at least 12 months before study randomisation.
The primary endpoint for the study was met at the planned interim analysis conducted after observing 80% of targeted progression-free survival (PFS) events using Response Evaluation Criteria in Solid Tumours (RECIST v1.1), based on the investigator assessment in the full population (all randomised patients), and confirmed by a blinded independent central radiological assessment.
The efficacy results demonstrated a statistically significant improvement in PFS in patients receiving Kisqali plus letrozole compared to patients receiving placebo plus letrozole in the full analysis set (hazard ratio of 0.556, 95% CI: 0.429, 0.720, one sided stratified log-rank test p-value 0.00000329) with clinically meaningful treatment effect.
The global health status/QoL data showed no relevant difference between the Kisqali plus letrozole arm and the placebo plus letrozole arm.
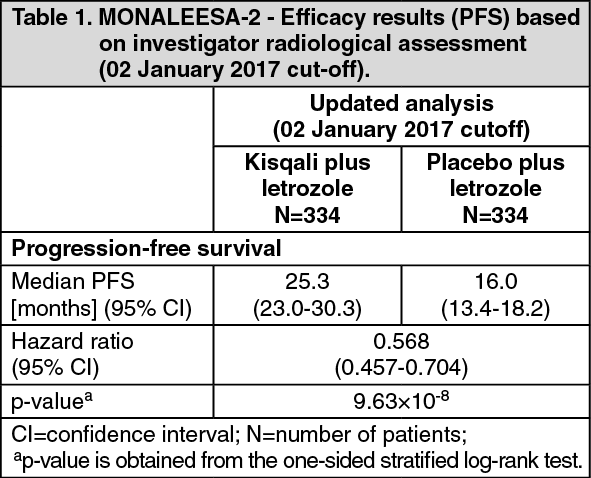
A more mature update of efficacy data (02 January 2017 cut-off) is provided in Tables 1 and 2.
Median PFS was 25.3 months (95% CI: 23.0, 30.3) for ribociclib plus letrozole treated patients and 16.0 months (95% CI: 13.4, 18.2) for patients receiving placebo plus letrozole. 54.7% of patients receiving ribociclib plus letrozole were estimated to be progression-free at 24 months compared with 35.9% in the placebo plus letrozole arm.
There was no statistically significant difference in overall survival (OS) between the Kisqali plus letrozole arm and the placebo plus letrozole arm (HR 0.746 [95% CI: 0.517, 1.078]). OS data remain immature. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image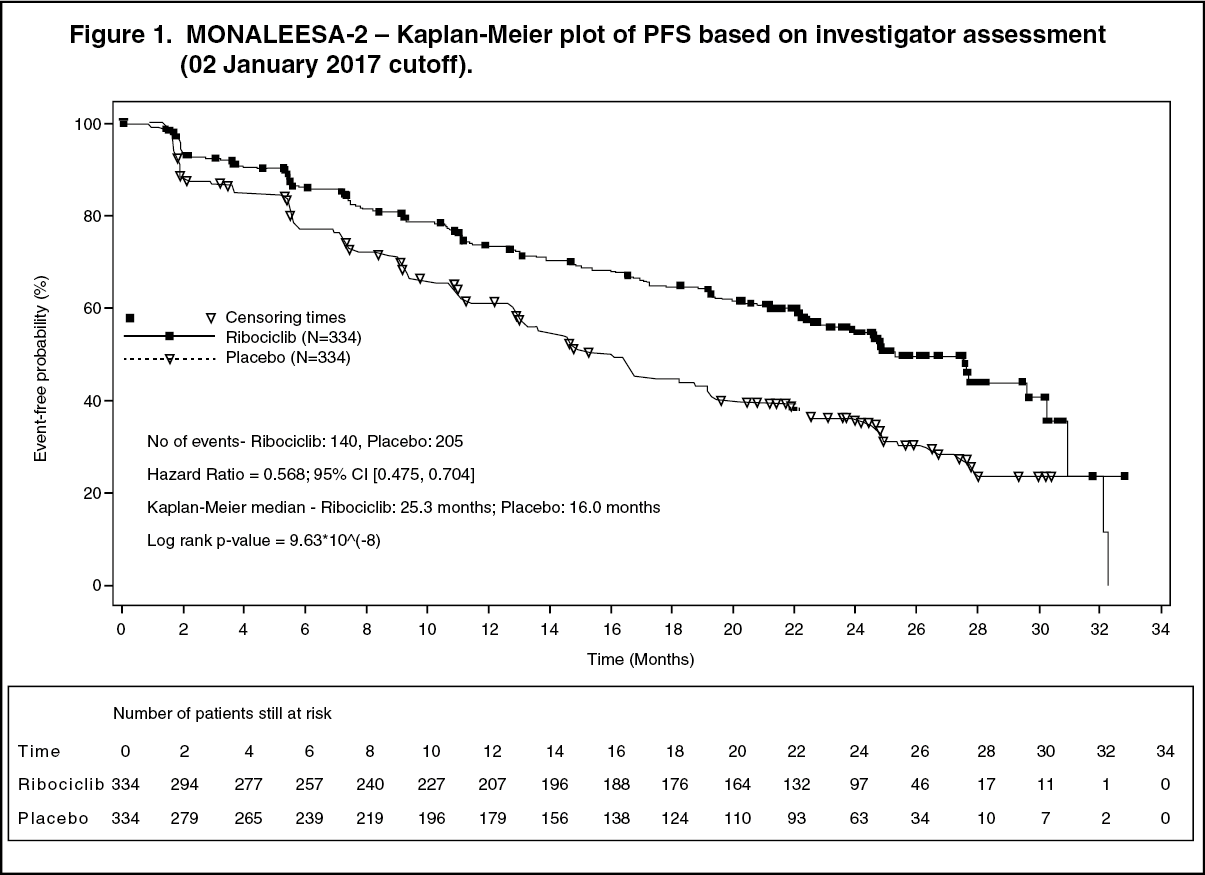 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA series of pre-specified subgroup PFS analyses was performed based on prognostic factors and baseline characteristics to investigate the internal consistency of treatment effect. A reduction in the risk of disease progression or death in favour of the Kisqali plus letrozole arm was observed in all individual patient subgroups of age, race, prior adjuvant or neoadjuvant chemotherapy or hormonal therapies, liver and/or lung involvement and bone-only metastatic disease. This was evident for patients with liver and/or lung metastases (HR of 0.561 [95% CI: 0.424, 0.743], median progression-free survival [mPFS] 24.8 months for Kisqali plus letrozole versus 13.4 months for letrozole alone), or without liver and/or lung metastases (HR of 0.597 [95% CI: 0.426, 0.837], mPFS 27.6 months versus 18.2 months).
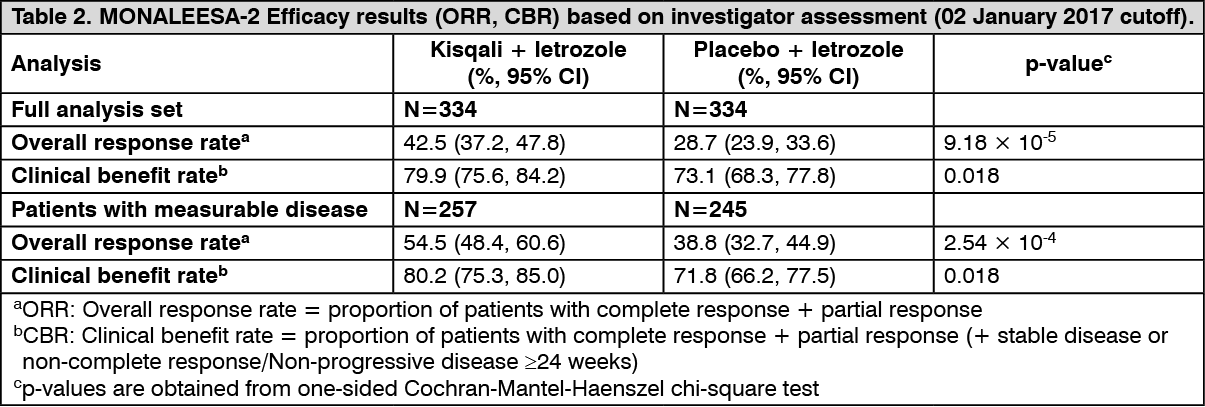
Updated results for overall response and clinical benefit rates are displayed in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageStudy CLEE011E2301 (MONALEESA-7): Kisqali was evaluated in a randomised, double-blind, placebo-controlled, multicentre phase III clinical study in the treatment of pre- and perimenopausal women with hormone receptor-positive, HER2-negative advanced breast cancer in combination with a NSAI or tamoxifen plus goserelin versus placebo in combination with a NSAI or tamoxifen plus goserelin. Patients in MONALEESA-7 had not received prior endocrine treatment in the advanced breast cancer setting.
A total of 672 patients were randomised in a 1:1 ratio to receive either Kisqali 600 mg plus NSAI/tamoxifen plus goserelin (n=335) or placebo plus NSAI/tamoxifen plus goserelin (n=337), stratified according to: the presence of liver and/or lung metastases (Yes [n=344 (51.2%)] versus No [n=328 (48.8%)]), prior chemotherapy for advanced disease (Yes [n=120 (17.9%)] versus No [n=552 (82.1%)]), and endocrine combination partner (NSAI and goserelin [n=493 (73.4%)] versus tamoxifen and goserelin [n=179 (26.6%)]). Demographics and baseline disease characteristics were balanced and comparable between study arms. Kisqali was given orally at a dose of 600 mg daily for 21 consecutive days followed by 7 days off treatment in combination with NSAI (letrozole 2.5 mg or anastrozole 1 mg) or tamoxifen (20 mg) orally once daily for 28 days, and goserelin (3.6 mg) subcutaneously every 28 days, until disease progression or unacceptable toxicity. Patients were not allowed to cross over from placebo to Kisqali during the study or after disease progression. Switching the endocrine combination partners was also not permitted.
Patients enrolled in this study had a median age of 44 years (range 25 to 58) and 27.7% of patients were younger than 40 years old. The majority of patients included were Caucasian (57.7%), Asian (29.5%) or Black (2.8%) and nearly all patients (99.0%) had a baseline ECOG performance status of 0 or 1. Prior to study entry, of these 672 patients, 14% of patients had received prior chemotherapy for metastatic disease, 32.6% of patients had received chemotherapy in the adjuvant and 18.0% in the neoadjuvant setting; 39.6% had received endocrine therapy in the adjuvant setting and 0.7% in the neoadjuvant setting. In study E2301 40.2% of patients had de novo metastatic disease, 23.7% had bone-only disease, and 56.7% had visceral disease.
The study met the primary endpoint at the primary analysis conducted after 318 progression-free survival (PFS) events based on the investigator assessment using RECIST v1.1 criteria in the full analysis set (all randomised patients). The primary efficacy results were supported by PFS results based on blinded independent central radiological assessment. The median follow-up time at the time of primary PFS analysis was 19.2 months.
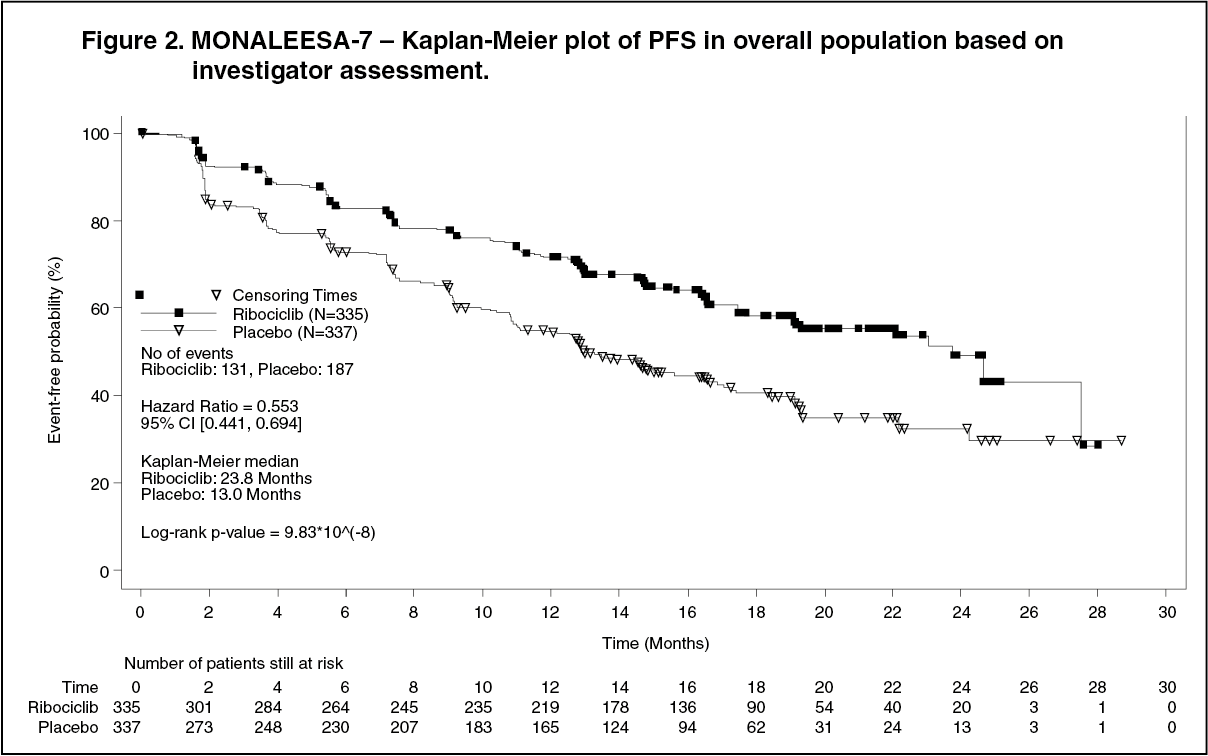
In the overall study population, the efficacy results demonstrated a statistically significant improvement in PFS in patients receiving Kisqali plus NSAI/tamoxifen plus goserelin compared to patients receiving placebo plus NSAI/tamoxifen plus goserelin (hazard ratio of 0.553, 95% CI: 0.441, 0.694, one-sided stratified log-rank test p-value 9.83x10-8) with clinically meaningful treatment effect. Median PFS was 23.8 months (95% CI: 19.2, NE) for Kisqali plus NSAI/tamoxifen plus goserelin treated patients and 13.0 months (95% CI: 11.0, 16.4) for patients receiving placebo plus NSAI/tamoxifen plus goserelin.
Distribution of PFS is summarised in the Kaplan-Meier curve for PFS in Figure 2. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe results for PFS based on the blinded independent central radiological assessment of a randomly selected subset of approximately 40% of randomised patients were supportive of the primary efficacy results based on the investigator's assessment (hazard ratio of 0.427; 95% CI: 0.288, 0.633).
At the time of the primary PFS analysis overall survival, data were not mature with 89 (13%) of events (N=252, HR 0.916 [95% CI: 0.601, 1.396]).
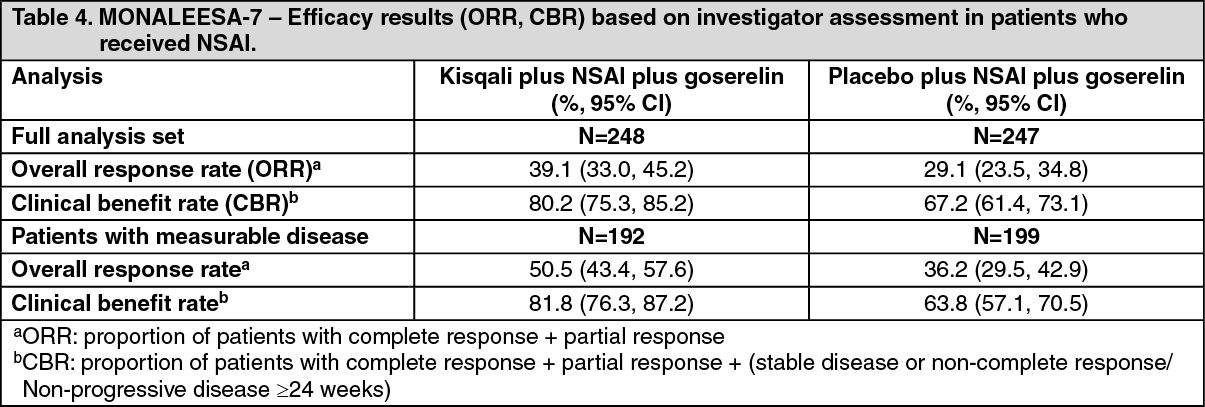
Overall response rate (ORR) per investigator assessment based on RECIST v1.1 was higher in the Kisqali arm (40.9%; 95% CI: 35.6, 46.2) compared to the placebo arm (29.7%; 95% CI: 24.8, 34.6, p=0.00098). The observed clinical benefit rate (CBR) was higher in Kisqali arm (79.1%; 95% CI: 74.8:83.5) compared to placebo arm (69.7%; 95% CI: 64.8:74.6, p=0.002).
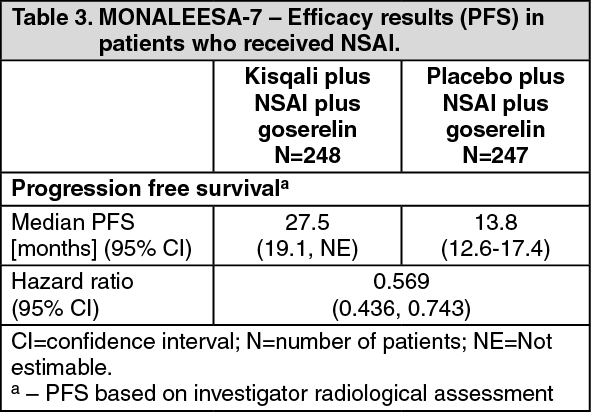
In the pre-specified subgroup analysis of 495 patients who had received Kisqali or placebo in combination with NSAI plus goserelin, the median PFS was 27.5 months (95% CI: 19.1, NE) in the Kisqali plus NSAI subgroup and 13.8 months (95% CI: 12.6, 17.4) in the placebo plus NSAI subgroup [HR: 0.569; 95% CI: 0.436, 0.743]. Efficacy results are summarised in Table 3 and the Kaplan-Meier curves for PFS are provided in Figure 3. (See Table 3 and Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image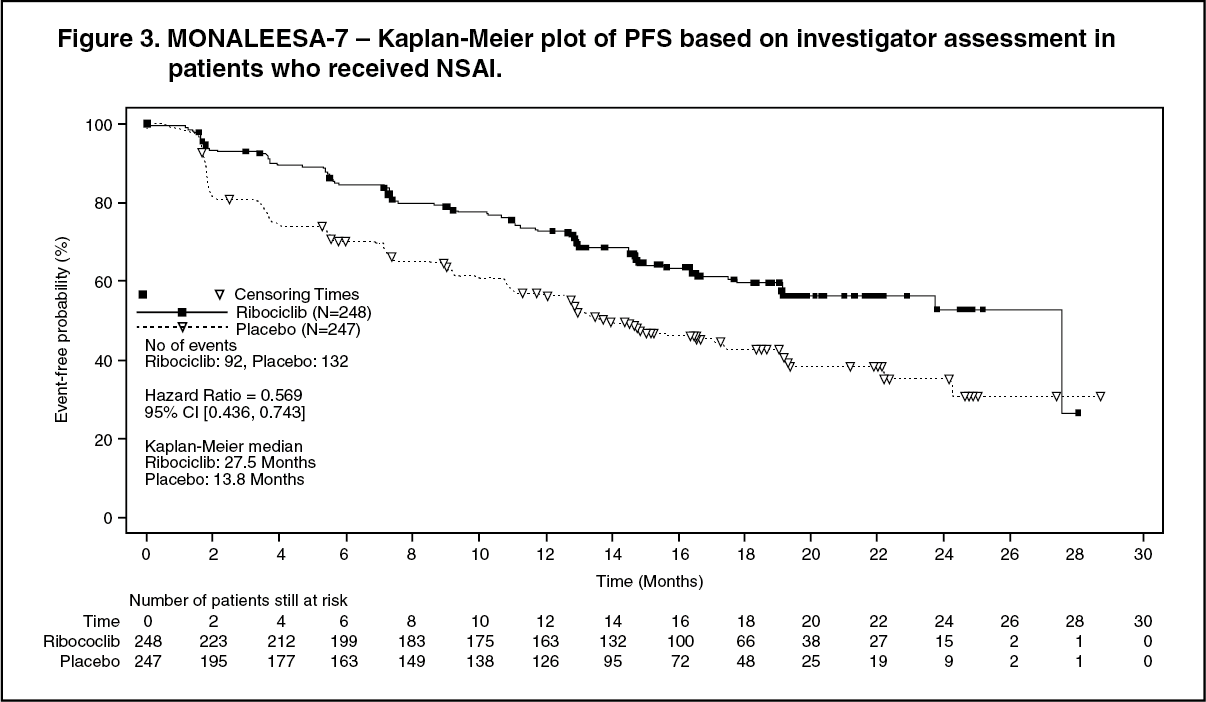 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEfficacy results for overall response rate (ORR) and clinical benefit rate (CBR) per investigator assessment based on RECIST v1.1 are provided in Table 4. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResults in the Kisqali plus NSAI subgroup were consistent across subgroups of age, race, prior adjuvant/neoadjuvant chemotherapy or hormonal therapies, liver and/or lung involvement and bone-only metastatic disease.
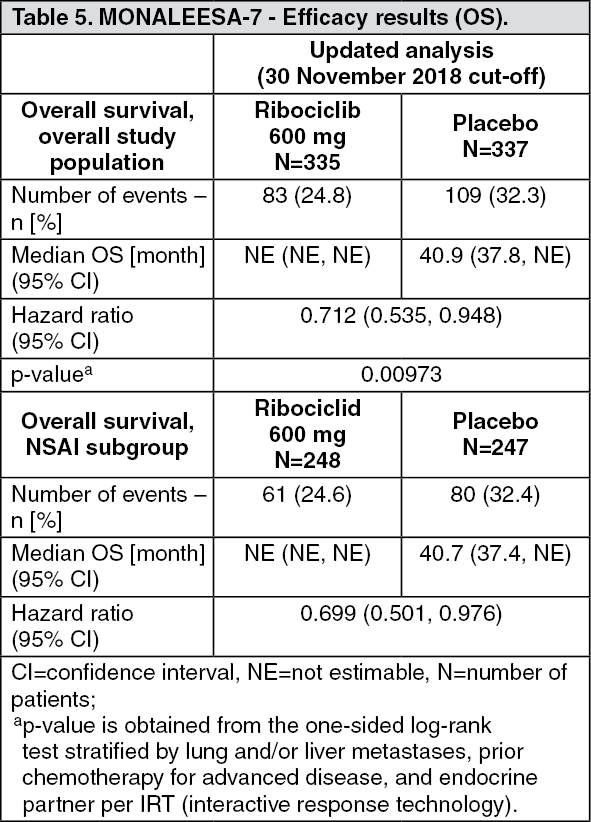
A more mature update of overall survival data (30 November 2018 cut-off) is provided in Table 5 and Figures 4 and 5.
In the second OS analysis the study met its key secondary endpoint demonstrating a statistically significant improvement in OS. (See Table 5 and Figures 4 and 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image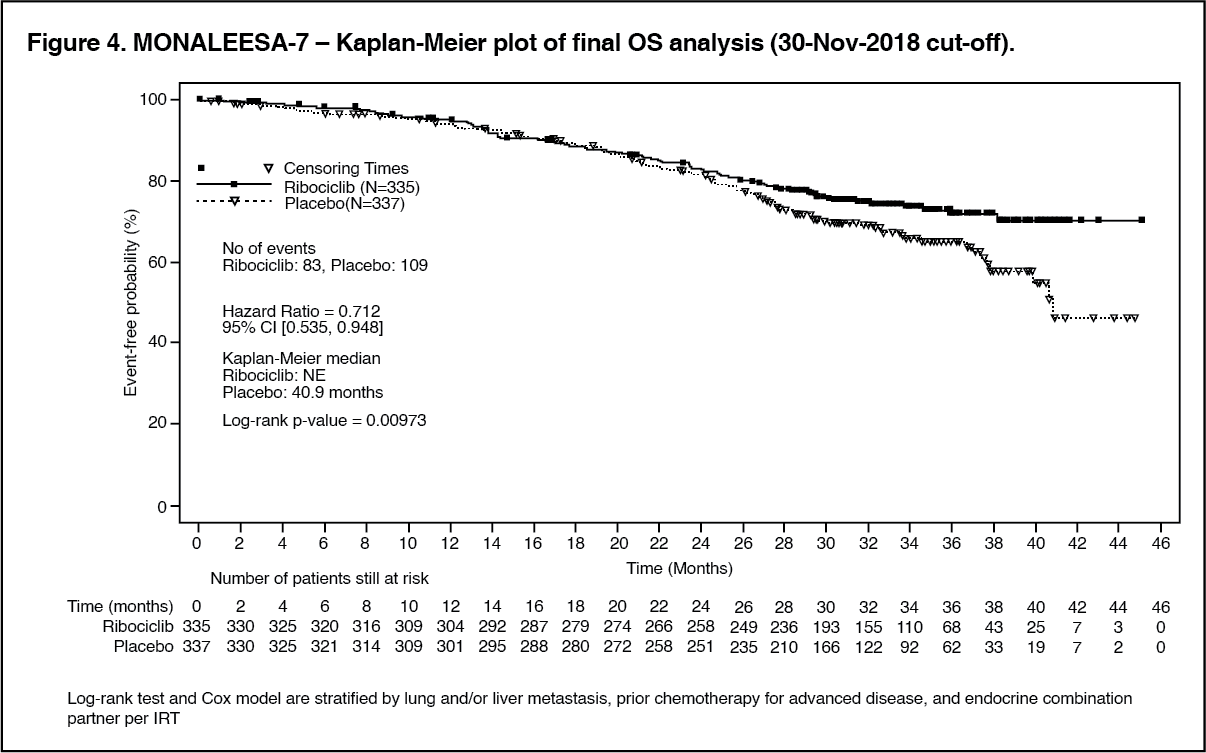 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image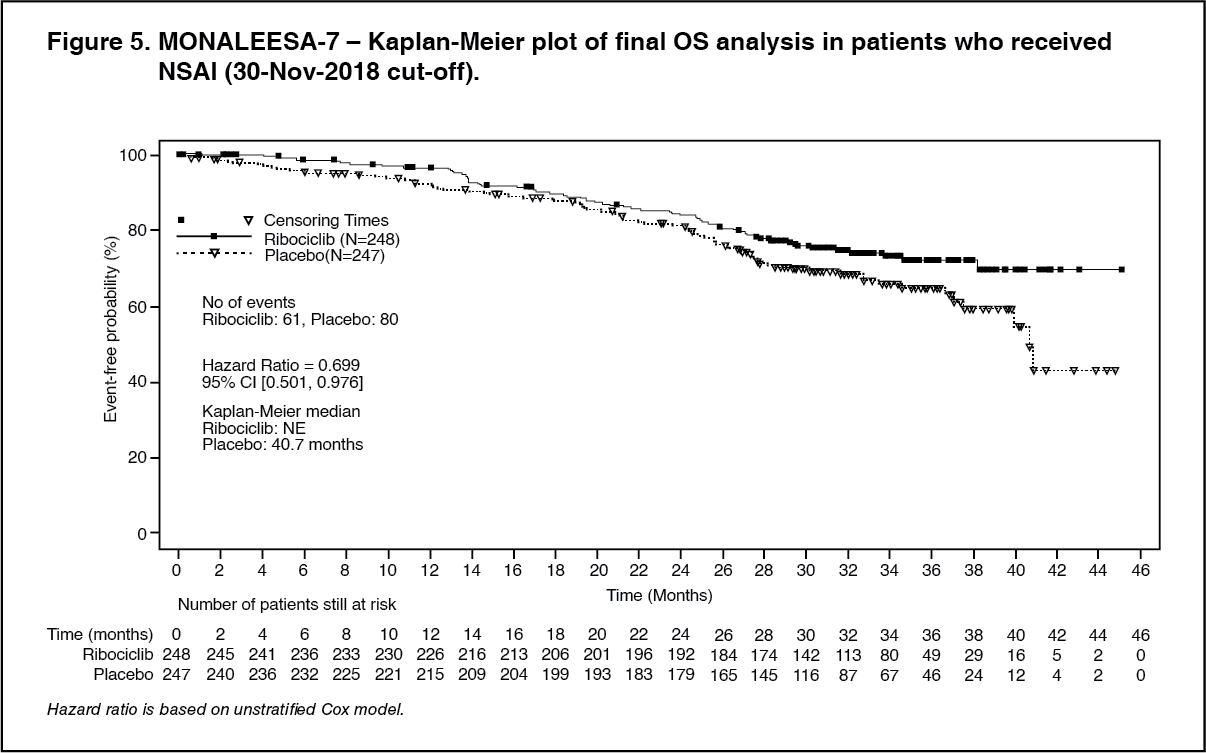 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditionally, the probability of progression on next-line therapy or death (PFS2) in patients who received prior ribociclib in the study was lower compared to patients in the placebo arm with an HR of 0.692 (95% CI: 0.548, 0.875) in the overall study population. The median PFS2 was 32.3 months (95% CI: 27.6, 38.3) in the placebo arm and was not reached (95% CI: 39.4, NE) for the ribociclib arm. Similar results were observed for the NSAI subgroup, with an HR of 0.660 (95% CI: 0.503, 0.868) and a median PFS2 of 32.3 months (95% CI: 26.9, 38.3) in the placebo arm versus not reached (95% CI: 39.4, NE) in the ribociclib arm.
Study CLEE011F2301 (MONALEESA-3): Kisqali was evaluated in a 2:1 randomised double-blind, placebo-controlled, multicentre phase III clinical study in 726 postmenopausal women with hormone receptor-positive, HER2-negative advanced breast cancer who had received no or only one line of prior endocrine treatment, in combination with fulvestrant versus fulvestrant alone.
Patients enrolled in this study had a median age of 63 years (range 31 to 89). 46.7% of patients were of age 65 years and older, including 13.8% patients of age 75 years and older. The patients included were Caucasian (85.3%), Asian (8.7%) or Black (0.7%) and nearly all patients (99.7%) had an ECOG performance status of 0 or 1. First and second line patients were enrolled in this study (of whom 19.1% had de novo metastatic disease). Prior to study entry 42.7% of patients had received chemotherapy in the adjuvant and 13.1% in the neoadjuvant setting, while 58.5% had received endocrine therapy in the adjuvant and 1.4% in the neoadjuvant setting and 21% had received prior endocrine therapy in the advanced breast cancer setting. In study F2301 21.2% had bone-only disease and 60.5% had visceral disease.
Primary analysis: The study met the primary endpoint at the primary analysis conducted after 361 progression-free survival (PFS) events based on the investigator assessment and using RECIST v1.1 criteria in the full analysis set (all randomised patients, 03 November 2017 cut-off). The median follow-up time at the time of primary PFS analysis was 20.4 months.
The primary efficacy results demonstrated a statistically significant improvement in PFS in patients receiving Kisqali plus fulvestrant compared to patients receiving placebo plus fulvestrant in the full analysis set (hazard ratio of 0.593, 95% CI: 0.480, 0.732, one-sided stratified log-rank test p-value 4.1x10-7), with an estimated 41% reduction in relative risk of progression or death in favour of the Kisqali plus fulvestrant arm.
The primary efficacy results were supported by a random central audit of 40% imaging subset by a blinded independent central radiological assessment (hazard ratio of 0.492; 95% CI: 0.345, 0.703).
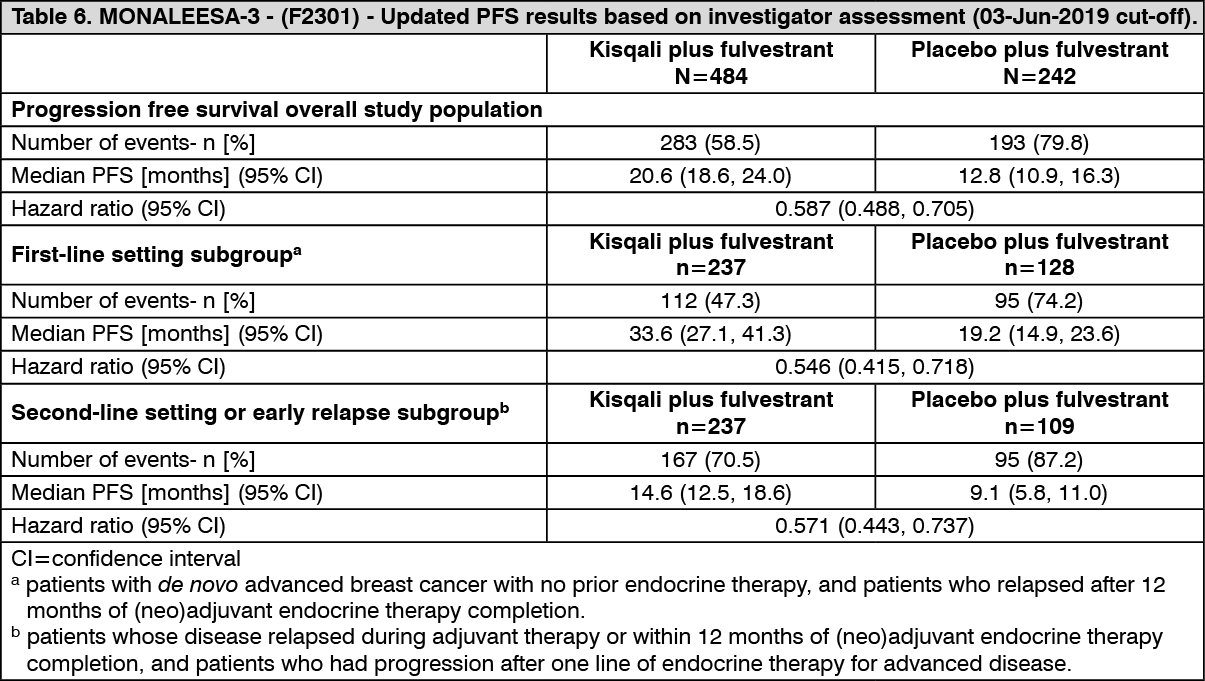
A descriptive update of PFS was performed at the time of the second OS interim analysis, and the updated PFS results on the overall population and the subgroups based on prior endocrine therapy are summarised in Table 6 and the Kaplan-Meier curve is provided in Figure 6. (See Table 6 and Figure 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image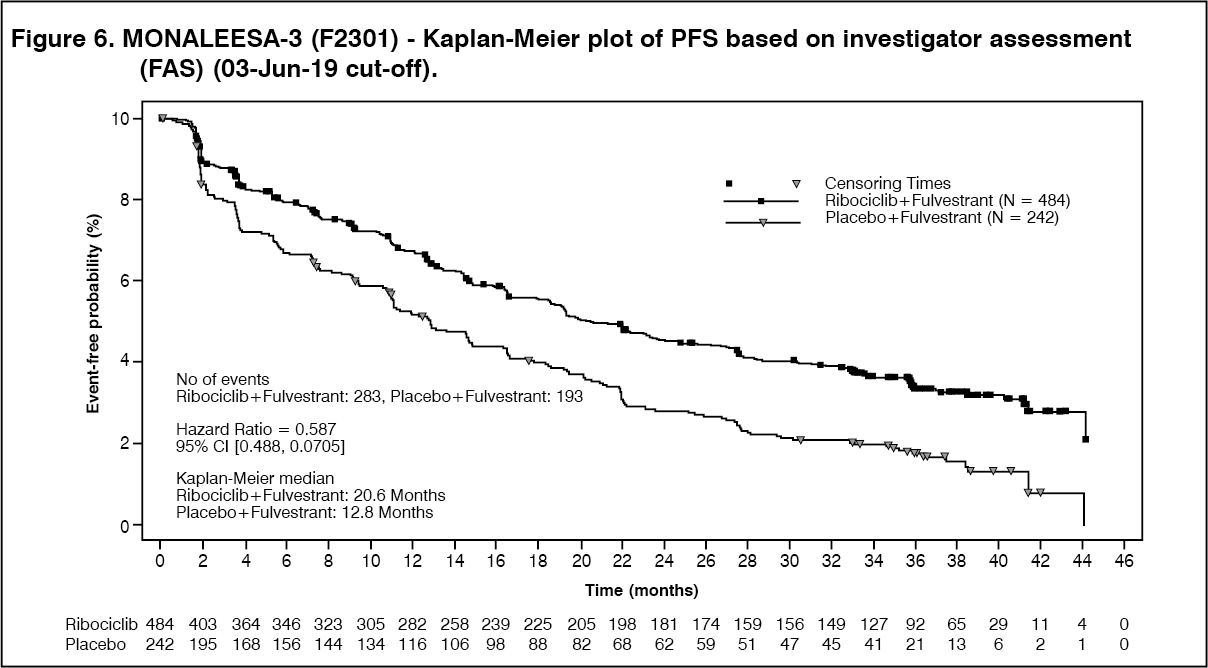 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEfficacy results for overall response rate (ORR) and clinical benefit rate (CBR) per investigator assessment based on RECIST v1.1 are provided in Table 7. (See Table 7.)
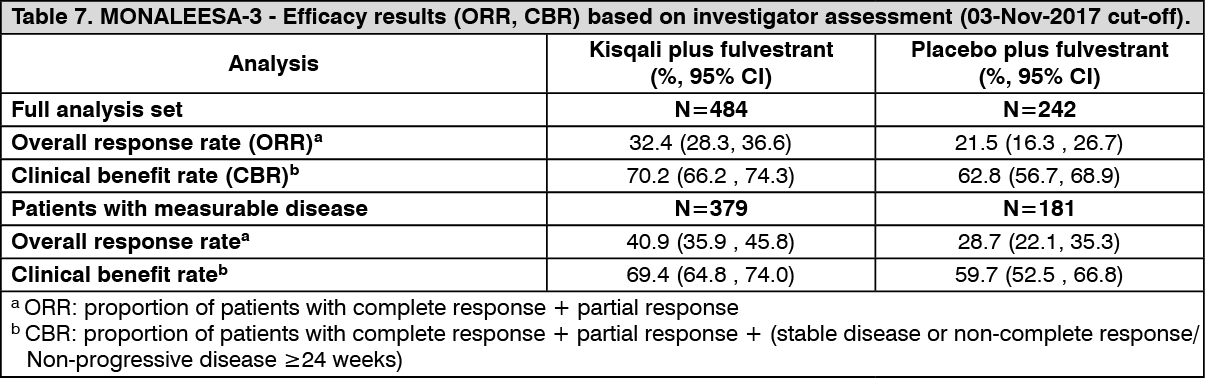 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageHazard ratios based on pre-specified subgroup analysis of the patients treated with Kisqali plus fulvestrant showed consistent benefit across different subgroups including age, prior treatment (early or advanced), prior adjuvant/neoadjuvant chemotherapy or hormonal therapies, liver and/or lung involvement and bone-only metastatic disease.
OS Analysis: In the pre-specified second OS interim analysis, the study met its secondary endpoint, demonstrating a statistically significant improvement in OS.
The results from this final OS analysis on the overall study population and the subgroups analysis are provided in Table 8 and Figure 7. (See Table 8 and Figure 7.)
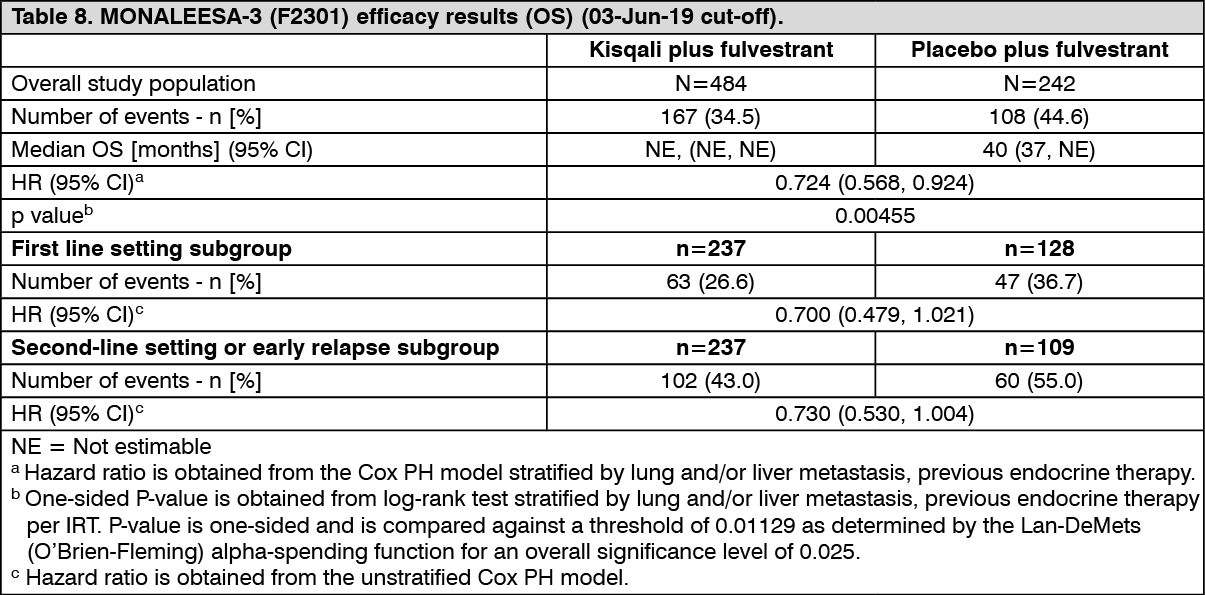 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image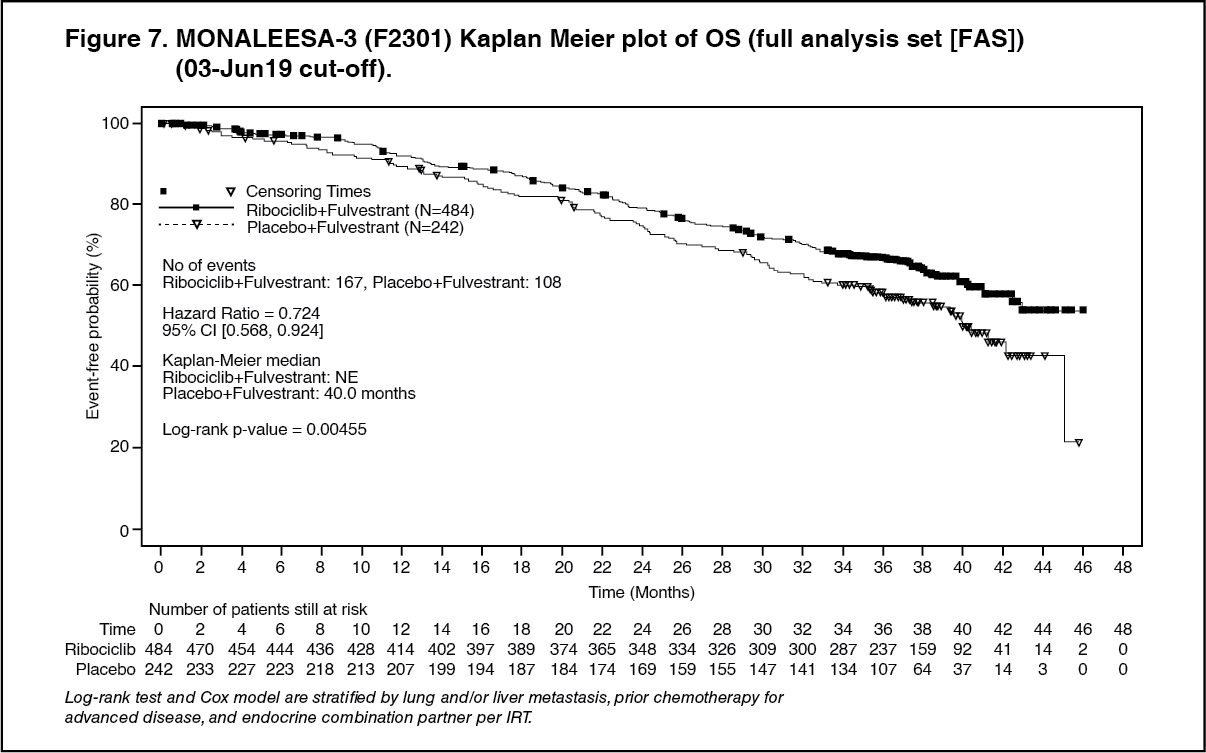 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTime to progression on next-line therapy or death (PFS2) in patients in the Kisqali arm was longer compared to patients in the placebo arm (HR: 0.670 [95% CI: 0.542, 0.830]) in the overall study population. The median PFS2 was 39.8 months (95% CI: 32.5, NE) for the Kisqali arm and 29.4 months (95% CI: 24.1, 33.1) in the placebo arm.
Study CLEE011A2404 (COMPLEEMENT-1): Kisqali was evaluated in an open-label, single arm, multicenter phase IIIb clinical study comparing ribociclib in combination with letrozole in pre/post-menopausal women and men with HR-positive, HER2-negative, advanced breast cancer who received no prior hormonal therapy for advanced disease. Premenopausal women, and men, also received goserelin or leuprolide.
The study enrolled 3246 patients, including 39 male patients who received Kisqali 600 mg orally once daily for 21 consecutive days followed by 7 days off; and letrozole 2.5 mg orally once daily for 28 days; and goserelin 3.6 mg as injectable subcutaneous implant or leuprolide 7.5 mg as intramuscular injection administered on Day 1 of each 28 day cycle. Patients were treated until disease progression or unacceptable toxicity occurred.
Male patients enrolled in this study had a median age of 62 years (range 33 to 80). Of these patients, 38.5% were 65 years and older, including 10.3% aged 75 years and older. The male patients enrolled were Caucasian (71.8%), Asian (7.7%), and Black (2.6%), with 17.9% unknown. Nearly all male patients (97.4%) had an ECOG performance status of 0 or 1. The majority of male patients (97%) had 4 or less metastatic sites, which were primarily bone and visceral (69.2% each). Table 9 summarizes the efficacy results in male patients. (See Table 9.)
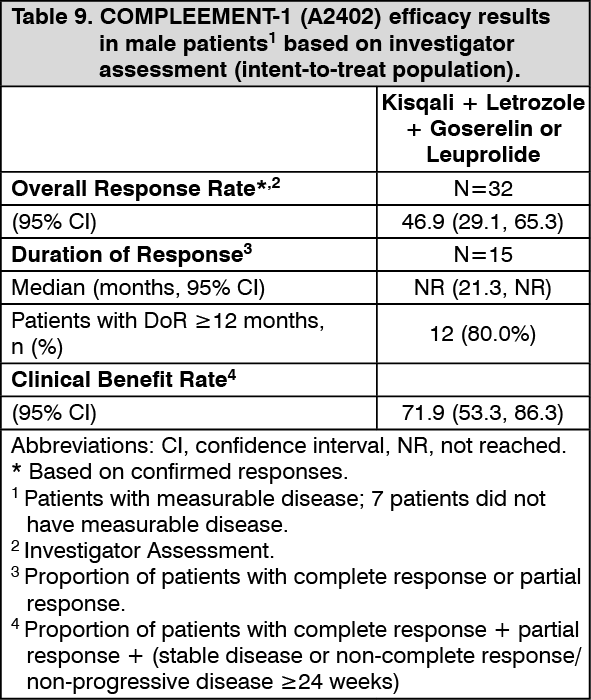 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageElderly patients: Of all patients who received Kisqali in studies MONALEESA-2 and MONALEESA-3, representative proportions of patients were ≥65 years and ≥75 years of age (see Pharmacodynamics as previously mentioned). No overall differences in safety or effectiveness of Kisqali were observed between these patients and younger patients (see Dosage & Administration).
Patients with renal impairment: In the three pivotal studies (MONALEESA-2, MONALEESA-3 and MONALEESA-7), 510 (53.8%) patients with normal renal function, 341 (36%) patients with mild renal impairment and 97 (10.2%) patients with moderate renal impairment were treated with ribociclib. No patient with severe renal impairment was enrolled. PFS results were consistent in patients with mild and moderate renal impairment who received ribociclib at the starting dose of 600 mg as compared to those with normal renal function. The safety profile was generally consistent across renal cohorts (see Adverse Reactions).
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with Kisqali in all subsets of the paediatric population in breast cancer (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: The pharmacokinetics of ribociclib were investigated in patients with advanced cancer following oral daily doses of 50 mg to 1200 mg. Healthy subjects received single oral doses ranging from 400 mg to 600 mg or repeated daily doses (8 days) at 400 mg.
Absorption: The absolute bioavailability of ribociclib is not known.
The time to reach Cmax (Tmax) following ribociclib oral administration was between 1 and 4 hours. Ribociclib exhibited slightly over-proportional increases in exposure (Cmax and AUC) across the dose range tested (50 to 1200 mg). Following repeated once-daily dosing, steady state was generally achieved after 8 days and ribociclib accumulated with a geometric mean accumulation ratio of 2.51 (range: 0.97 to 6.40).
Food effect: Compared to the fasted state, oral administration of a single 600 mg dose of ribociclib film-coated tablets with a high-fat, high-calorie meal had no effect on the rate and extent of absorption of ribociclib.
Distribution: Binding of ribociclib to human plasma proteins in vitro was approximately 70% and was independent of concentration (10 to 10000 ng/ml). Ribociclib was equally distributed between red blood cells and plasma with a mean in vivo blood-to-plasma ratio of 1.04. The apparent volume of distribution at steady state (Vss/F) was 1090 L based on population pharmacokinetic analysis.
Biotransformation: In vitro and in vivo studies indicated ribociclib is eliminated primarily via hepatic metabolism mainly via CYP3A4 in humans. Following oral administration of a single 600 mg dose of [14C] ribociclib to humans, the primary metabolic pathways for ribociclib involved oxidation (dealkylation, C and/or N-oxygenation, oxidation (-2H)) and combinations thereof. Phase II conjugates of ribociclib phase I metabolites involved N-acetylation, sulfation, cysteine conjugation, glycosylation and glucuronidation. Ribociclib was the major circulating drug-derived entity in plasma. The major circulating metabolites included metabolite M13 (CCI284, N-hydroxylation), M4 (LEQ803, N-demethylation), and M1 (secondary glucuronide). Clinical activity (pharmacological and safety) of ribociclib was due primarily to parent drug, with negligible contribution from circulating metabolites.
Ribociclib was extensively metabolised, with unchanged drug accounting for 17.3% and 12.1% of the dose in faeces and urine, respectively. Metabolite LEQ803 was a significant metabolite in excreta and represented approximately 13.9% and 3.74% of the administered dose in faeces and urine, respectively. Numerous other metabolites were detected in both faeces and urine in minor amounts (≤2.78% of the administered dose).
Elimination: The geometric mean plasma effective half-life (based on accumulation ratio) was 32.0 hours (63% CV) and the geometric mean apparent oral clearance (CL/F) was 25.5 l/hr (66% CV) at steady state at 600 mg in patients with advanced cancer. The geometric mean apparent plasma terminal half-life (T½) of ribociclib ranged from 29.7 to 54.7 hours and the geometric mean CL/F of ribociclib ranged from 39.9 to 77.5 l/hr at 600 mg across studies in healthy subjects.
Ribociclib and its metabolites are eliminated mainly via faeces, with a small contribution of the renal route. In 6 healthy male subjects, following a single oral dose of [14C] ribociclib, 91.7% of the total administered radioactive dose was recovered within 22 days; faeces was the major route of excretion (69.1%), with 22.6% of the dose recovered in urine.
Linearity/non-linearity: Ribociclib exhibited slightly over-proportional increases in exposure (Cmax and AUC) across the dose range of 50 mg to 1200 mg following both single dose and repeated doses. This analysis is limited by the small sample sizes for most of the dose cohorts with a majority of the data coming from the 600 mg dose cohort.
Special populations: Renal impairment: The effect of renal function on the pharmacokinetics of ribociclib was assessed in a renal impairment study that included 14 healthy subjects with normal renal function (absolute Glomerular Filtration Rate [aGFR] ≥90 ml/min), 8 subjects with mild renal impairment (aGFR 60 to <90 ml/min), 6 subjects with moderate renal impairment (aGFR 30 to <60 ml/min), 7 subjects with severe renal impairment (aGFR 15 to <30 ml/min) and 3 subjects with end-stage renal disease (ESRD) (aGFR <15 ml/min) at a single ribociclib dose of 400 mg.
AUCinf increased 1.6-fold, 1.9-fold and 2.7-fold and Cmax increased 1.8-fold, 1.8-fold and 2.3-fold in subjects with mild, moderate and severe renal impairment, relative to the exposure in subjects with normal renal function. Since the efficacy and safety studies of ribociclib included a large proportion of patients with mild renal impairment (see Pharmacodynamics as previously mentioned), data from the subjects with moderate or severe renal impairment in the renal impairment study were also compared with pooled data for the subjects with normal renal function and mild renal impairment. Compared to the pooled data for the subjects with normal renal function and mild renal impairment, AUCinf increased 1.6-fold and 2.2-fold and Cmax increased 1.5-fold and 1.9-fold in subjects with moderate and severe renal impairment, respectively. A fold difference for subjects with ESRD was not calculated due to the small number of subjects, but results indicate a similar or somewhat larger increase in ribociclib exposure compared to subjects with severe renal impairment.
The effect of renal function on the pharmacokinetics of ribociclib was also assessed in cancer patients included in efficacy and safety studies where patients were given the 600 mg start dose (see Pharmacodynamics as previously mentioned). In a sub-group analysis of pharmacokinetic data from studies in cancer patients following oral administration of 600 mg ribociclib as a single dose or repeat doses, AUCinf and Cmax of ribociclib in patients with mild (n=57) or moderate (n=14) renal impairment were comparable to the AUCinf and Cmax in patients with normal renal function (n=86) suggesting no clinically meaningful effect of mild or moderate renal impairment on ribociclib exposure.
Hepatic impairment: Based on a pharmacokinetic study in non-cancer subjects with hepatic impairment, mild hepatic impairment had no effect on the exposure of ribociclib (see Dosage & Administration). The mean exposure for ribociclib was increased less than 2-fold in patients with moderate (geometric mean ratio [GMR]: 1.44 for Cmax; 1.28 for AUCinf) and severe (GMR: 1.32 for Cmax; 1.29 for AUCinf) hepatic impairment (see Dosage & Administration).
Based on a population pharmacokinetic analysis that included 160 breast cancer patients with normal hepatic function and 47 patients with mild hepatic impairment, mild hepatic impairment had no effect on the exposure of ribociclib, further supporting the findings from the dedicated hepatic impairment study. Ribociclib has not been studied in breast cancer patients with moderate or severe hepatic impairment.
Effect of age, weight, gender and race: Population pharmacokinetic analysis showed that there are no clinically relevant effects of age, body weight or gender on the systemic exposure of ribociclib that would require a dose adjustment. Data on differences in pharmacokinetics due to race are too limited to draw conclusions.
In vitro interaction data: Effect of ribociclib on cytochrome P450 enzymes: In vitro, ribociclib is a reversible inhibitor of CYP1A2, CYP2E1 and CYP3A4/5 and a time-dependent inhibitor of CYP3A4/5, at clinically relevant concentrations. In vitro evaluations indicated that ribociclib has no potential to inhibit the activities of CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 at clinically relevant concentrations. Ribociclib has no potential for time-dependent inhibition of CYP1A2, CYP2C9, and CYP2D6.
In vitro data indicate that ribociclib has no potential to induce UGT enzymes or the CYP enzymes CYP2C9, CYP2C19 and CYP3A4 via PXR. Therefore, Kisqali is unlikely to affect substrates of these enzymes. In vitro data are not sufficient to exclude a potential of ribociclib to induce CYP2B6 via CAR.
Effect of transporters on ribociclib: Ribociclib is a substrate for P-gp in vitro, but based on mass balance data inhibition of P-gp or BCRP is unlikely to affect ribociclib exposure at therapeutic doses. Ribociclib is not a substrate for hepatic uptake transporters OATP1B1, OATP1B3 or OCT-1 in vitro.
Effect of ribociclib on transporters: In vitro evaluations indicated that ribociclib has a potential to inhibit the activities of drug transporters P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 and BSEP. Ribociclib did not inhibit OAT1, OAT3 or MRP2 at clinically relevant concentrations in vitro.
Toxicology: Preclinical safety data: Safety pharmacology: In vivo cardiac safety studies in dogs demonstrated dose and concentration related QTc interval prolongation at an exposure that would be expected to be achieved in patients following the recommended dose of 600 mg. There is also potential to induce incidences of premature ventricular contractions (PVCs) at elevated exposures (approximately 5-fold the anticipated clinical Cmax).
Repeated-dose toxicity: Repeated-dose toxicity studies (treatment schedule of 3 weeks on/1 week off) of up to 27 weeks' duration in rats and up to 39 weeks' duration in dogs, revealed the hepatobiliary system (proliferative changes, cholestasis, sand-like gallbladder calculi, and inspissated bile) as the primary target organ of toxicity of ribociclib. Target organs associated with the pharmacological action of ribociclib in repeat-dose studies include bone marrow (hypocellularity), lymphoid system (lymphoid depletion), intestinal mucosa (atrophy), skin (atrophy), bone (decreased bone formation), kidney (concurrent degeneration and regeneration of tubular epithelial cells) and testes (atrophy). Besides the atrophic changes seen in the testes, which showed a trend towards reversibility, all other changes were fully reversible after a 4-week treatment-free period. Exposure to ribociclib in animals in the toxicity studies was generally less than or equal to that observed in patients receiving multiple doses of 600 mg/day (based on AUC).
Reproductive toxicity/Fertility: Ribociclib showed foetotoxicity and teratogenicity at doses which did not show maternal toxicity in the rats or rabbits. Following prenatal exposure, increased incidences of post-implantation loss and reduced foetal weights were observed in rats and ribociclib was teratogenic in rabbits at exposures lower than or 1.5 times the exposure in humans, respectively, at the highest recommended dose of 600 mg/day based on AUC.
In rats, reduced foetal weights accompanied by skeletal changes considered to be transitory and/or related to the lower foetal weights were noted. In rabbits, there were adverse effects on embryo-foetal development as evidenced by increased incidences of foetal abnormalities (malformations and external, visceral and skeletal variants) and foetal growth (lower foetal weights). These findings included reduced/small lung lobes and additional vessel on the aortic arch and diaphragmatic hernia, absent accessory lobe or (partly) fused lung lobes and reduced/small accessory lung lobe (30 and 60 mg/kg), extra/rudimentary thirteenth ribs and misshapen hyoid bone and reduced number of phalanges in the pollex. There was no evidence of embryo-foetal mortality.
In a fertility study in female rats, ribociclib did not affect reproductive function, fertility or early embryonic development at any dose up to 300 mg/kg/day (which is likely at an exposure lower than or equal to patients' clinical exposure at the highest recommended dose of 600 mg/day based on AUC).
Ribociclib has not been evaluated in male fertility studies. However, atrophic changes in testes were reported in rat and dog toxicity studies at exposures that were less than or equal to human exposure at the highest recommended daily dose of 600 mg/day based on AUC. These effects can be linked to a direct anti-proliferative effects on the testicular germ cells resulting in atrophy of the seminiferous tubules.
Ribociclib and its metabolites passed readily into rat milk. The exposure to ribociclib was higher in milk than in plasma.
Genotoxicity: Genotoxicity studies in bacterial in vitro systems and in mammalian in vitro and in vivo systems with and without metabolic activation did not reveal any evidence for a genotoxic potential of ribociclib.
Carcinogenesis: Ribociclib was assessed for carcinogenicity in a 2-year study in rats.
Oral administration of ribociclib for 2 years resulted in an increased incidence of endometrial epithelial tumours and glandular and squamous hyperplasia in the uterus/cervix of female rats at doses ≥300 mg/kg/day as well as an increased incidence in follicular tumours in the thyroid glands of male rats at a dose of 50 mg/kg/day. Mean exposure at steady state (AUC0-24h) in female and male rats in whom neoplastic changes were seen was 1.2- and 1.4-fold that achieved in patients at the recommended dose of 600 mg/day, respectively. Mean exposure at steady state (AUC0-24h) in female and male rats in whom neoplastic changes were seen was 2.2- and 2.5-fold that achieved in patients at a dose of 400 mg/day, respectively.
Additional non-neoplastic proliferative changes consisted of increased liver altered foci (basophilic and clear cell) and testicular interstitial (Leydig) cell hyperplasia in male rats at doses of ≥5 mg/kg/day and 50 mg/kg/day, respectively.
The effects on the uterus/cervix and on the testicular interstitial (Leydig) cells may be related to prolonged hypoprolactinemia secondary to CDK4 inhibition of lactotrophic cell function in the pituitary gland, altering the hypothalamus-pituitary-gonadal axis.
Potential mechanisms for the thyroid findings in males include a rodent-specific microsomal enzyme induction in the liver and/or a dysregulation of the hypothalamus-pituitary-testis-thyroid axis secondary to a persistent on-target hypoprolactinemia.
Any potential increase of oestrogen/progesterone ratio in humans by this mechanism would be compensated by an inhibitory action of concomitant anti-oestrogen therapy on oestrogen synthesis as in humans Kisqali is indicated in combination with oestrogen-lowering agents.
Considering important differences between rodents and humans with regard to synthesis and role of prolactin, this mode of action is not expected to have consequences in humans.