


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Effect on Pruritus: This product inhibited a scratching behavior induced by intradermal administration of histamine in mice, a model wherein the existing antipruritics of antihistamines are effective, and the scratching behavior induced by intradermal administration of substance P in mice, a model wherein antihistamines are resistant. Moreover, a scratching behavior induced by intracisternal administration of morphine in mice, a central itching model wherein antihistamines are ineffective, was also inhibited by this product.
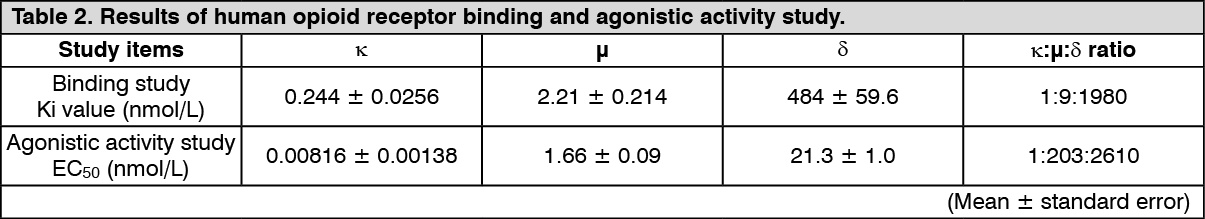
Mechanism of Action: This product has been shown to be a selective κ-opioid receptor agonist based on the results of in vitro receptor binding and agonistic activity study using human opioid receptor expressing cells. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn vitro studies have shown that this product does not bind to the various receptors and ion channels including histamine receptors other than the opioid receptors, and that this product does not inhibit the degranulation response in mast cells. Furthermore, the scratching inhibitory effect of this product in mice induced by intradermal administration of substance P was completely blocked by the intracerebroventricular administration of norbinaltorphimine (nor-BNI) which is an antagonist for κ-opioid receptor.
Dependence: The physical dependence liability of this product is considered to be weak because the withdrawal signs found by the termination of the repeated administration of morphine were barely shown by that of nalfurafine hydrochloride in rats. It is thought that there is no psychological dependence liability on this product because no reinforcing effect was observed on the self-administration study in monkeys.
Clinical studies: Results of nalfurafine hydrochloride (capsule) are shown as follows.
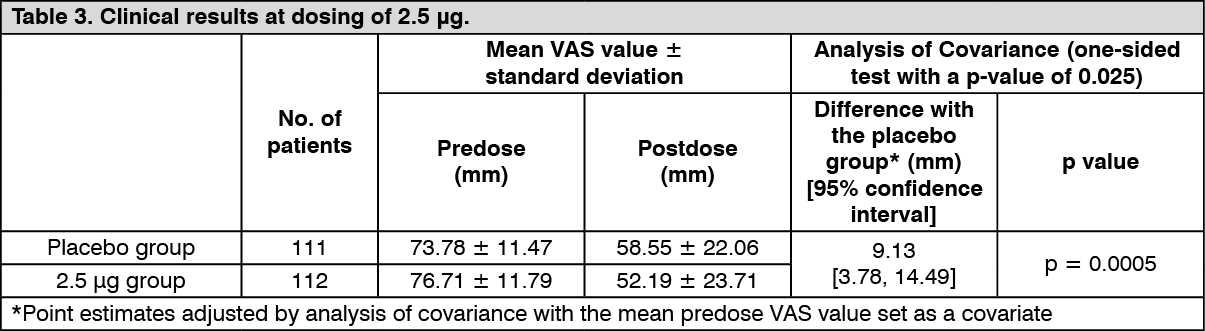
Improvement of pruritus in hemodialysis patients: Confirmatory study: The efficacy once daily repeated-dose oral administration once daily for 14 days was examined using a Visual Analogue Scale (VAS) which is an index of itching in a multicenter, double blind, comparative study in 337 hemodialysis patients who have pruritus refractory to the existing treatment(s). The efficacy, evaluated by the amount of change of VAS from predose to postdose compared to placebo, was confirmed in the 2.5 μg and 5 μg administration groups. (See Tables 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image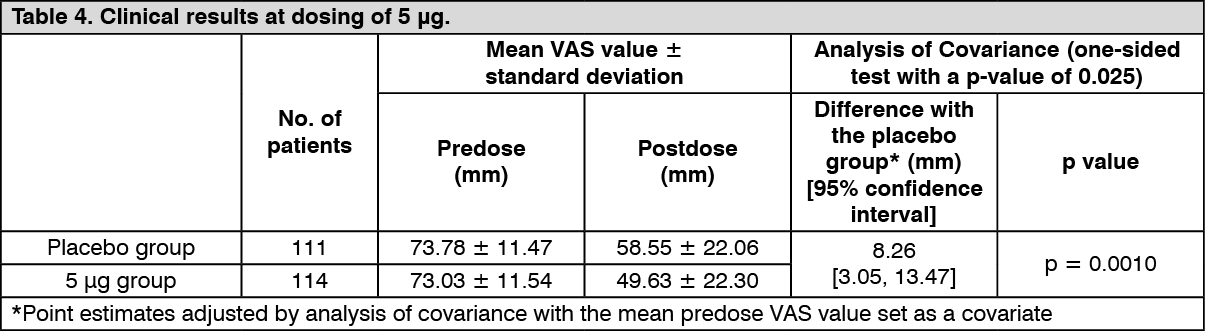 Click on icon to see table/diagram/image
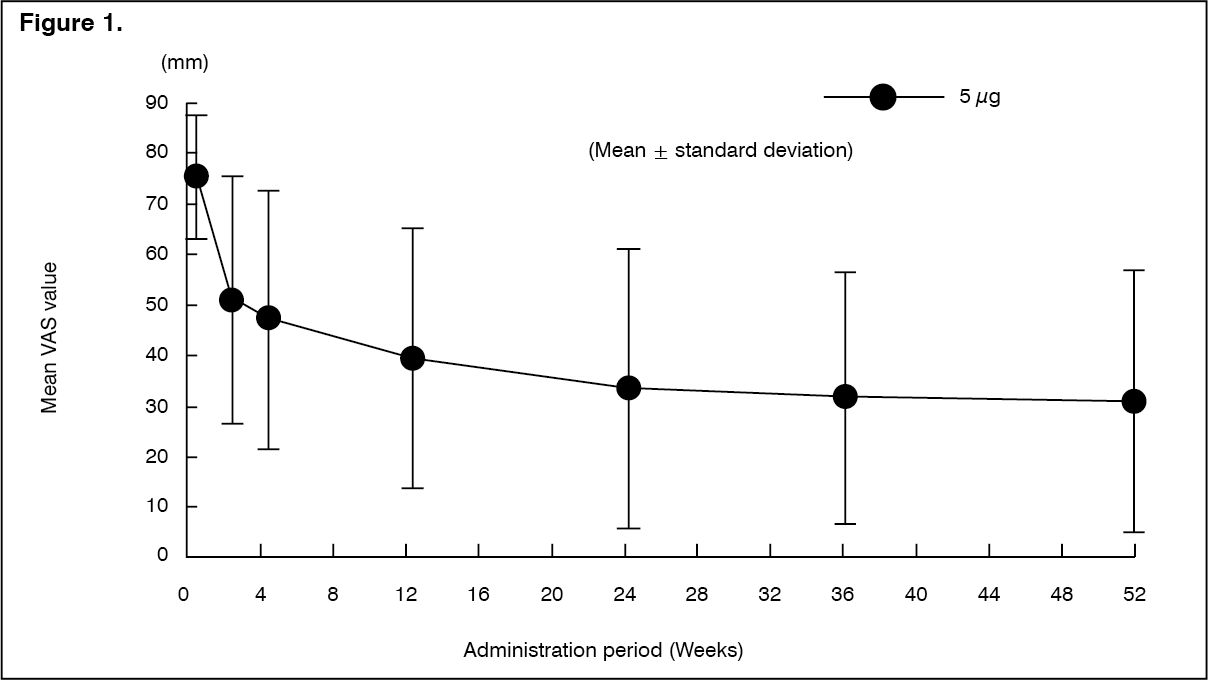
Click on icon to see table/diagram/imageLong-term administration study: The efficacy at the time of repeated-dose oral administration of nalfurafine hydrochloride 5 μg once daily for 52 weeks was examined using the VAS in an open-label study in 211 hemodialysis patients who had pruritus refractory to the existing treatment(s). The efficacy of this product, evaluated by the amount of change in VAS from baseline compared to after medication administration, was confirmed. (See Figure 1 and Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image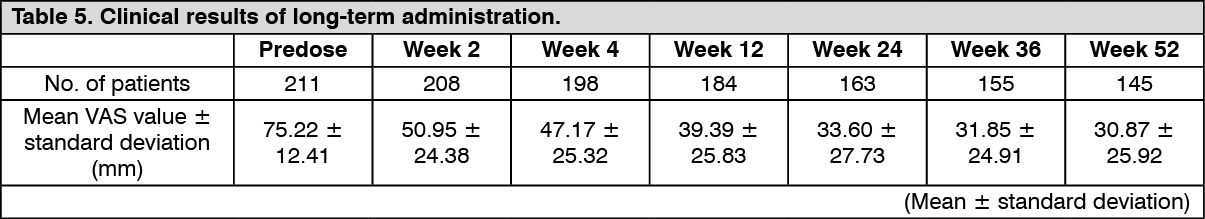 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNo patients showing a psychological or physical dependence on nalfurafine hydrochloride were observed. In addition, tolerance of efficacies was observed in 5 of 211 patients.
Improvement of pruritus in peritoneal dialysis patients: Uncontrolled open-label study: The efficacy of repeated-dose oral administration of nalfurafine hydrochloride (the dosage was 2.5 μg for 2 weeks and continuously increased to 5 μg for 2 weeks) was examined using a VAS, which is an index of itching, in an uncontrolled open-label study in 37 peritoneal dialysis patients who had pruritus refractory to the existing treatment(s). The mean change in VAS from baseline compared to after 2.5 μg administration for two weeks [LOCF: Last Observation Carried Forward] was 24.93 mm [18.67, 31.19] (90% CI), and the lower limit of the 90% CI for the mean change in VAS (18.67 mm) was greater than the threshold value (15.24 mm) which was set before the study.
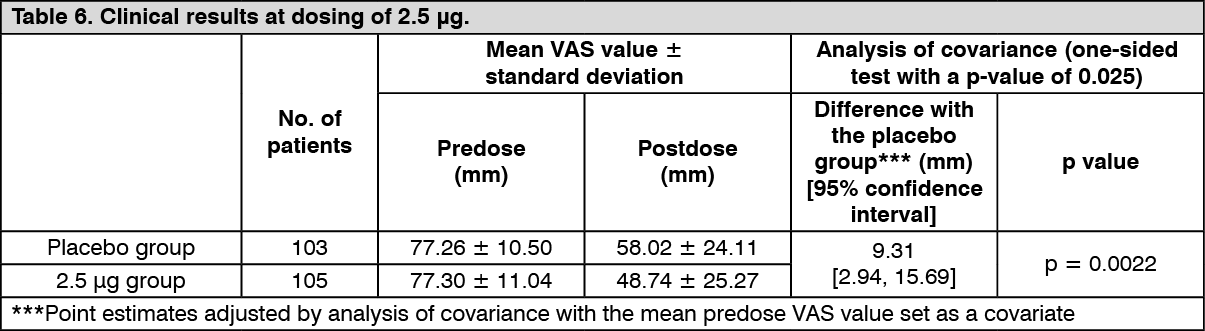
Improvement of pruritus in patients with chronic liver disease: Confirmatory study: The efficacy of repeated-dose oral administration once daily for 12 weeks was examined using a VAS, which is an index of itching, in a multicenter, double-blind, comparative study in 316 patients with chronic liver disease* who had pruritus refractory to antihistamine agents or antiallergic agents. The primary endpoint was the amount of change in VAS at Week 4 in the administration period (LOCF). The efficacy, evaluated by the amount of change in VAS from predose to postdose compared with placebo, was confirmed in the 2.5 μg and 5 μg administration groups.
*Patients with liver disease in whom the underlying disease has been confirmed, and the hepatic inflammation persists for at least 6 months or progression of disease state of hepatitis is confirmed by diagnostic imaging. (See Tables 6 and 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image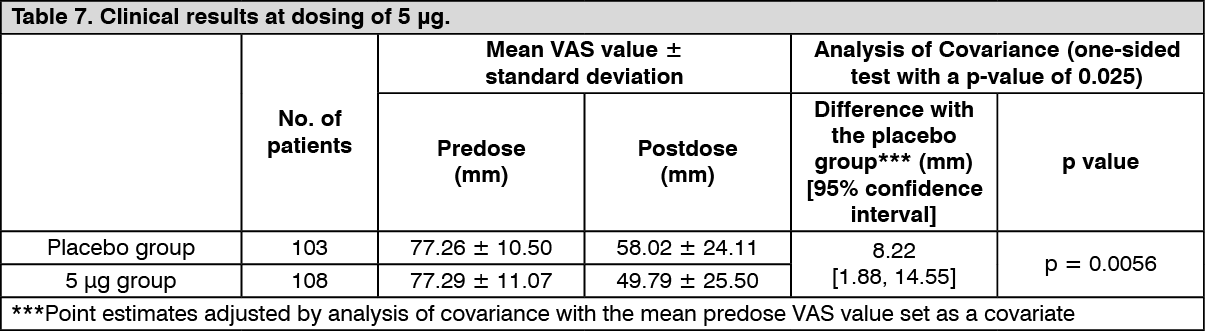 Click on icon to see table/diagram/image
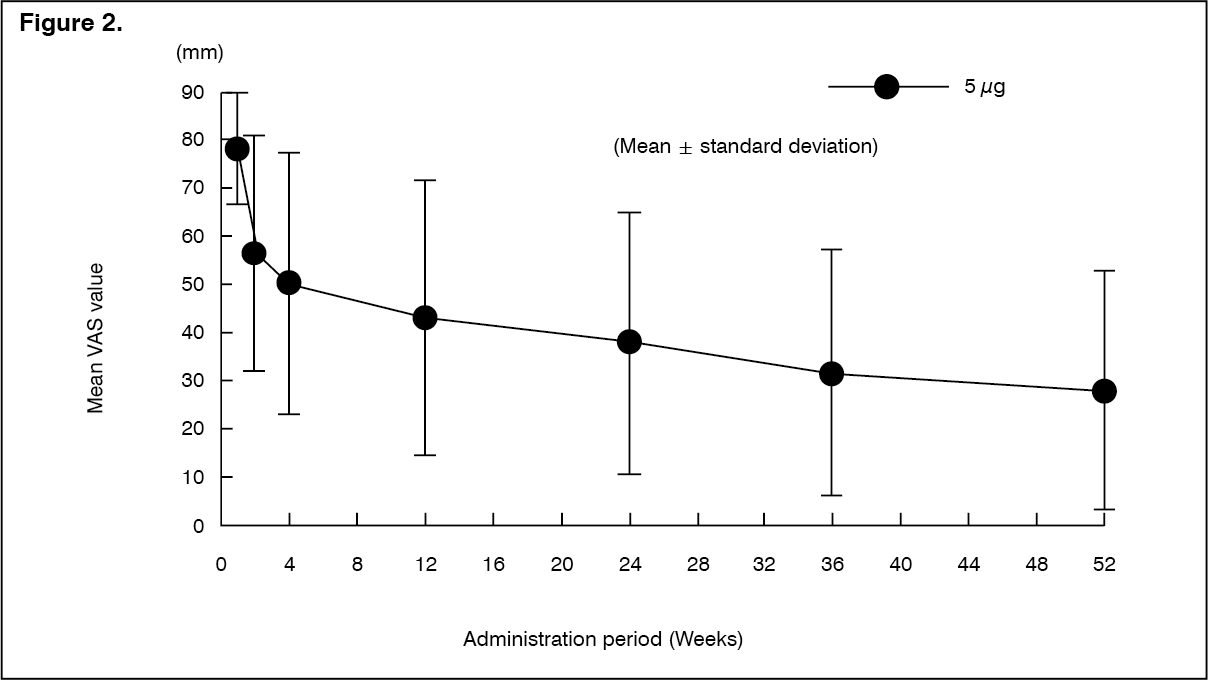
Click on icon to see table/diagram/imageLong-term administration study: The efficacy of repeated-dose oral administration of nalfurafine hydrochloride 5 μg once daily for 52 weeks was examined using the VAS in an open-label study in 122 patients with chronic liver disease* who had pruritus refractory to antihistamine agents or antiallergic agents. The efficacy of this product, evaluated by the amount of change in VAS from predose compared with postdose, was confirmed.
*Patients with liver disease in whom the underlying disease has been confirmed, and the hepatic inflammation persists for at least 6 months or progression of disease state of hepatitis is confirmed by diagnostic imaging. (See Figure 2 and Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image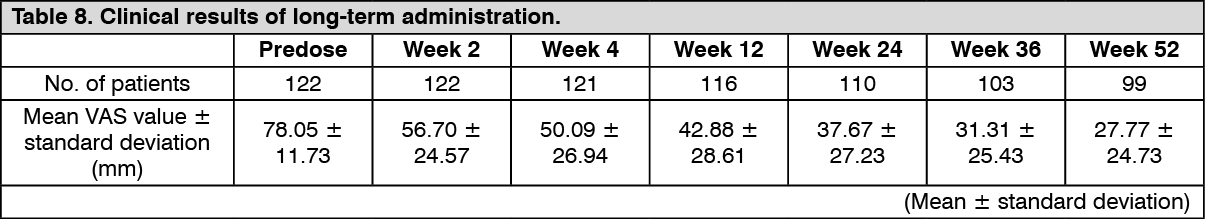 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNo patients showing a psychological dependence on nalfurafine hydrochloride were observed. In addition, physical dependence was observed in 1 and tolerance of efficacies was observed in 4 of 122 patients.
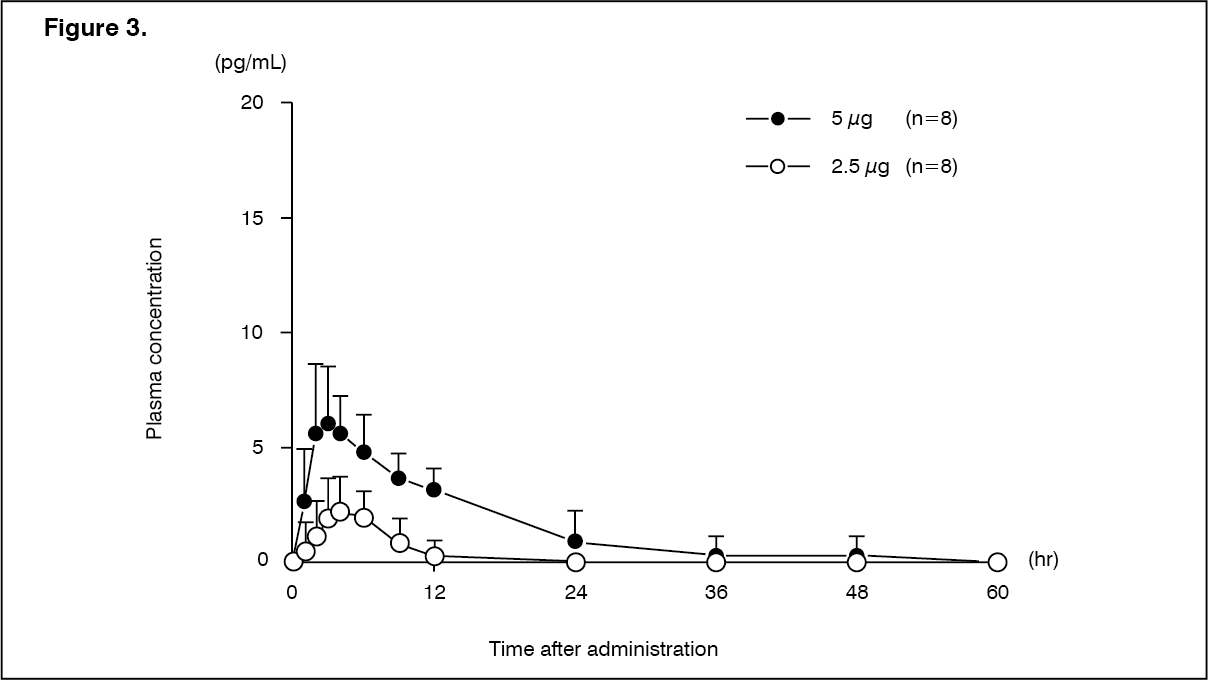
Pharmacokinetics: Absorption: Hemodialysis patients (single dose): Changes over time in the plasma concentration and the pharmacokinetic parameters of the unchanged drug after a single-dose oral administration of nalfurafine hydrochloride (capsule) 2.5 or 5 μg to 16 hemodialysis patients are shown as follows. (See Figure 3 and Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image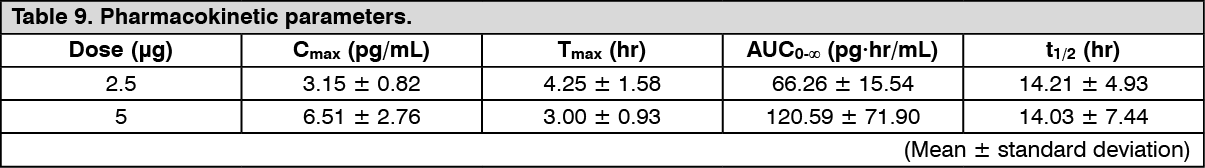 Click on icon to see table/diagram/image
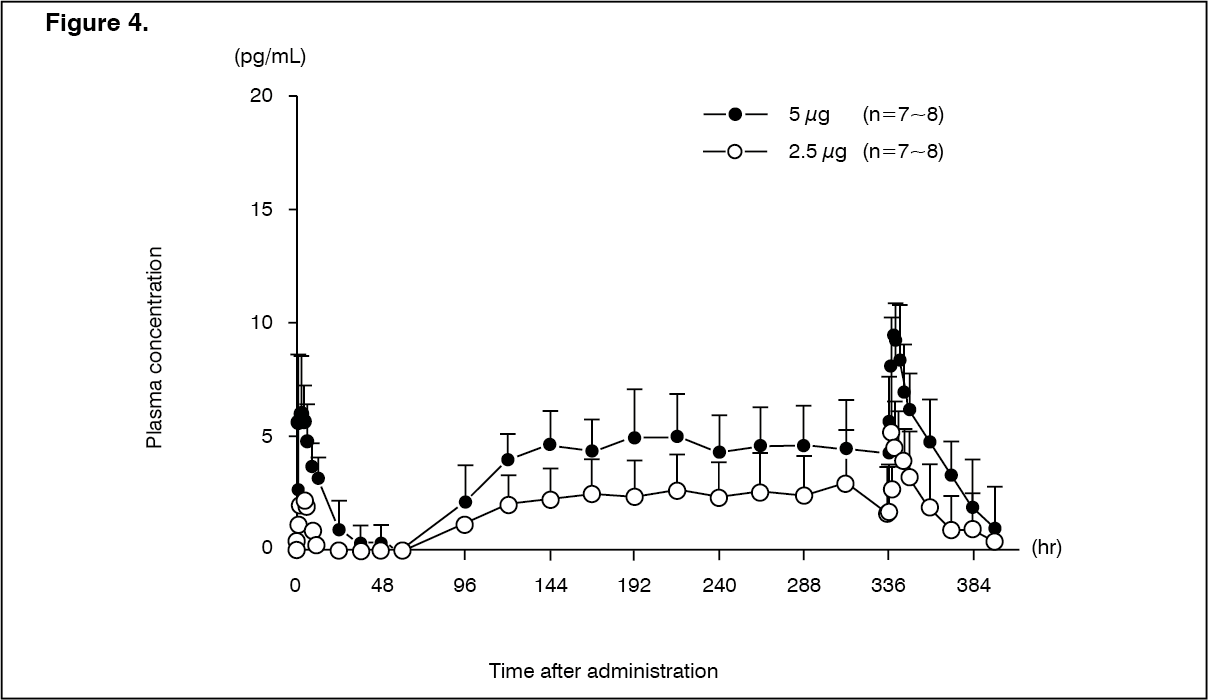
Click on icon to see table/diagram/imageHemodialysis patients (repeated dose): Changes over time in the plasma concentration and the pharmacokinetic parameters of the unchanged drug after repeated-dose oral administrations of nalfurafine hydrochloride (capsule) 2.5 or 5 μg to 14-16 hemodialysis patients are shown as follows. (See Figure 4 and Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image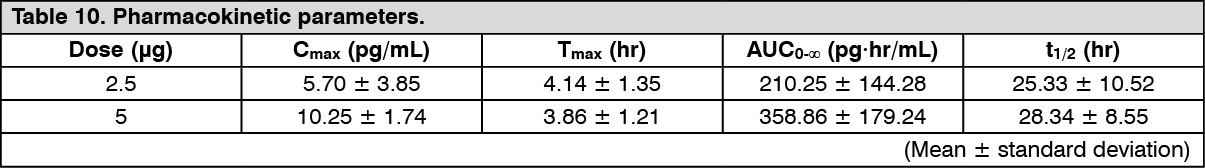 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt the time of dialysis, t½ was shortened when compared with that in the absence of dialysis. The t½ in the presence or absence of hemodialysis is 7.60 ± 2.02 hours and 32.06 ± 15.50 hours, respectively.
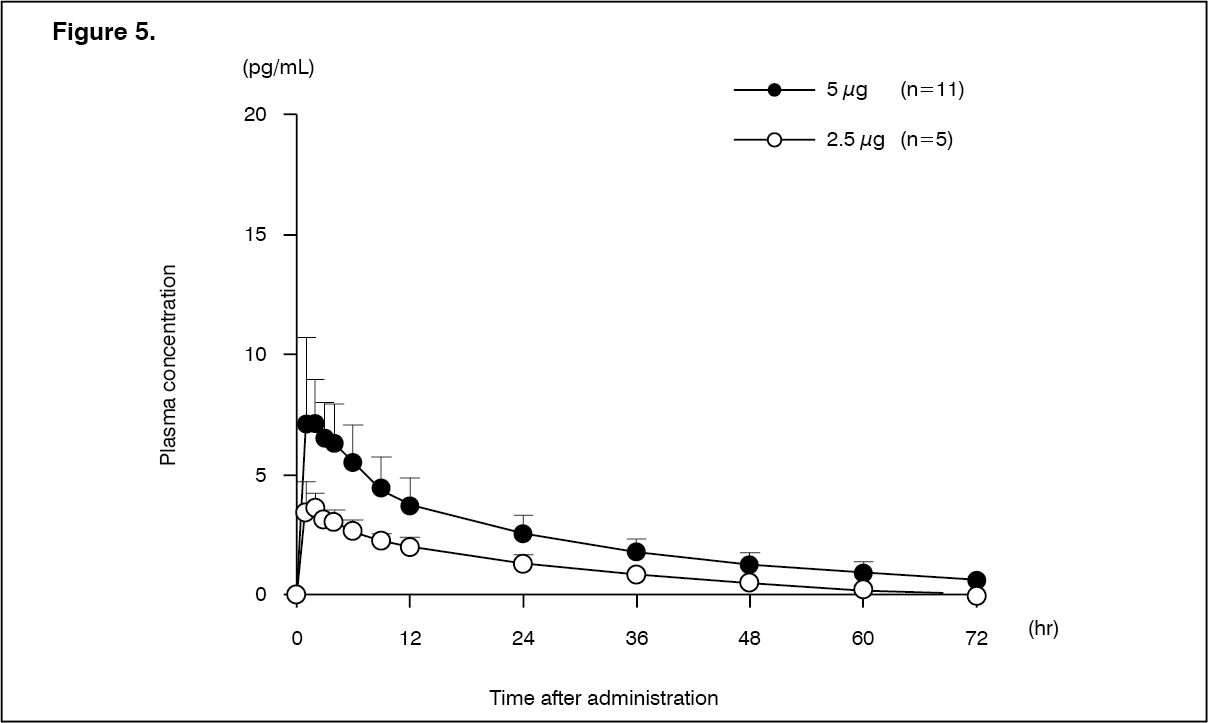
Peritoneal dialysis patients (single dose): Changes over time in the plasma concentration and the pharmacokinetic parameters of the unchanged drug after a single-dose oral administration of nalfurafine hydrochloride (capsule) 2.5 or 5 μg to 16 peritoneal dialysis patients are shown as follows. There was no apparent difference in pharmacokinetic parameters of the unchanged drug depending on the types of peritoneal dialysis [Continuous Ambulatory Peritoneal Dialysis (CAPD), Continuous Cycling Peritoneal Dialysis (CCPD)], use/non-use of Automated Peritoneal Dialysis (APD), and the types of dialysate solution. In 1 of 5 patients who received first dialysate exchange at 3 hours post-dose in the 5 μg administration group, there was a tendency of decreasing Cmax and AUC0-∞ of the unchanged drug to 5.37 pg/mL and 156.54 pg·hr/mL, respectively. (See Figure 5 and Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image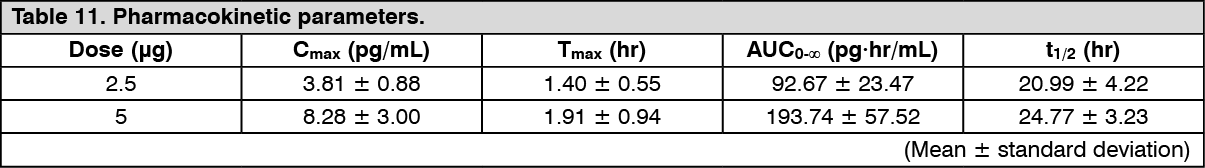 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients with mild hepatic dysfunction (Child-Pugh grade A): Changes over time in the plasma concentration and pharmacokinetic parameters of the unchanged drug after a single-dose oral administration of nalfurafine hydrochloride (capsule) 2.5 or 5 μg to 12 patients with compensated cirrhosis in Child-Pugh grade A are shown as follows. There was no tendency of increasing Cmax and AUC in comparison with healthy adult males. (See Figure 6 and Table 12.)
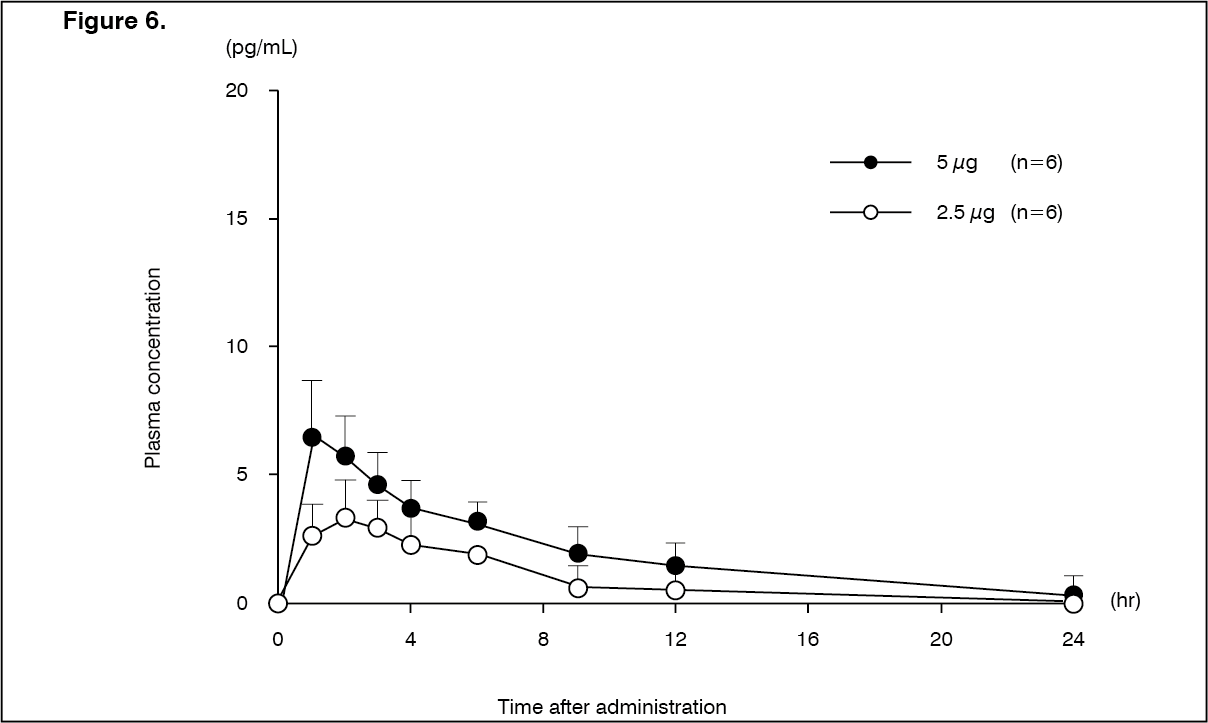 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image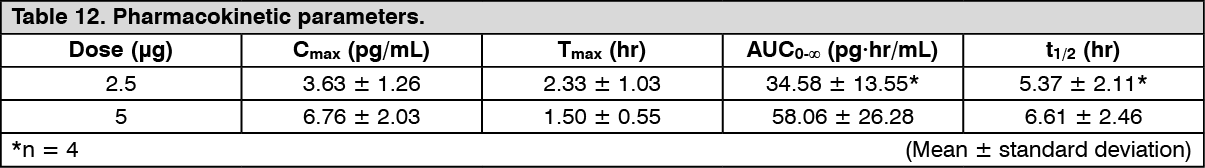 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients with moderate hepatic dysfunction (Child-Pugh grade B): Changes over time in the plasma concentration and pharmacokinetic parameters of the unchanged drug after a single-dose oral administration of nalfurafine hydrochloride (capsule) 2.5 or 5 μg to 30 patients with chronic hepatic disease in Child-Pugh grade B are shown as follows. There was a tendency of increasing Cmax and AUC in comparison with patients with mild hepatic dysfunction (Child-Pugh grade A). (See Figure 7 and Table 13.)
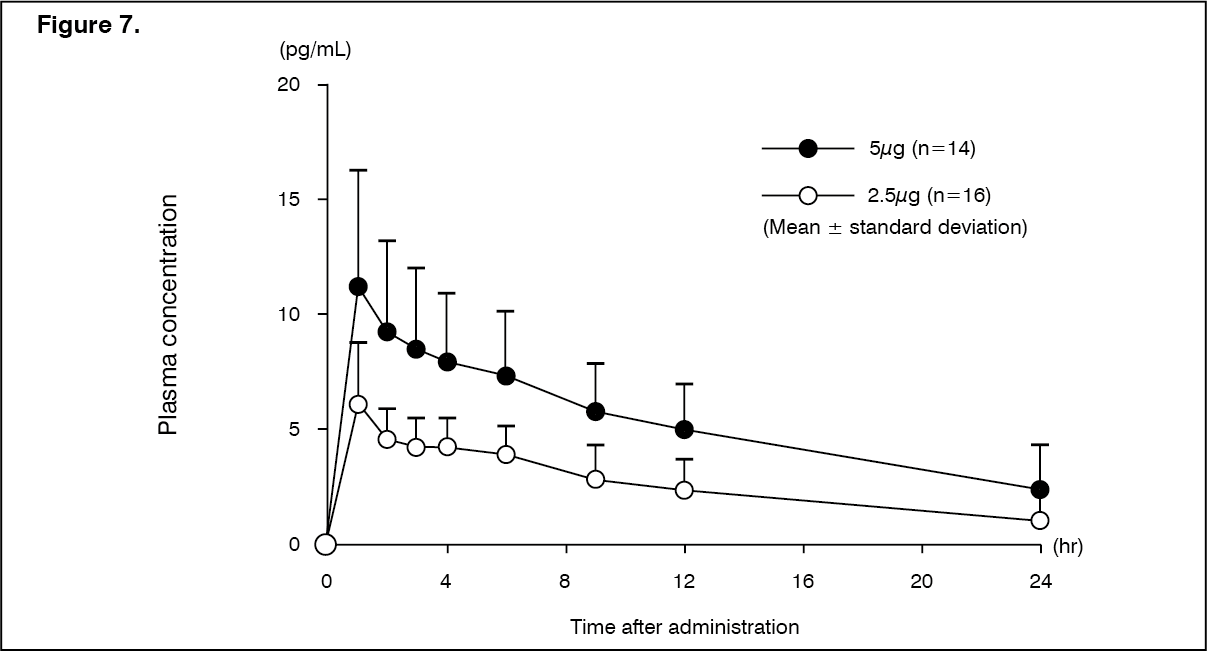 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image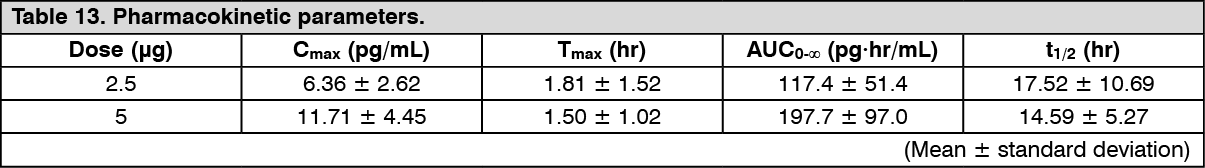 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients with severe hepatic dysfunction (Child-Pugh grade C): Pharmacokinetics in patients with hepatic dysfunction in Child-Pugh grade C has not been investigated.
Bioequivalence study: Changes over time in the plasma concentration and the pharmacokinetic parameters of the unchanged drug after a single-dose oral administration of nalfurafine hydrochloride (this product and capsule) 5 μg to 30 healthy male adults are shown as follows. The bioequivalence between the existing capsule formulation and this product (with and without water) formulation has been confirmed. (See Figure 8 and Table 14.)
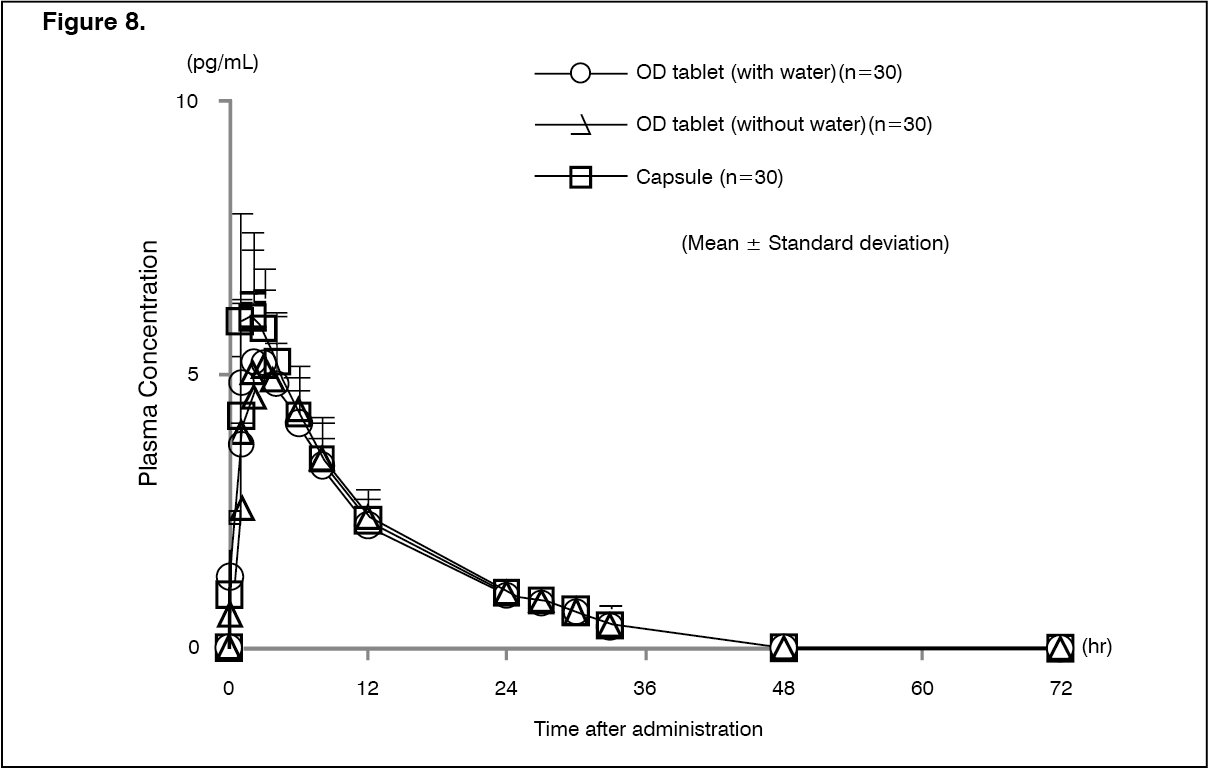 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image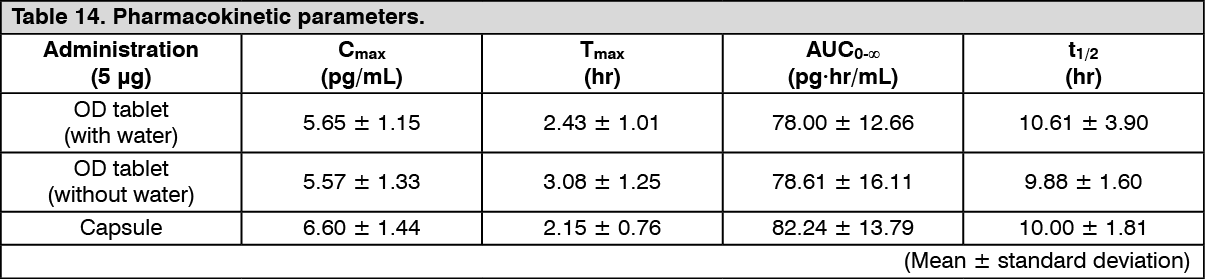 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDistribution: In vitro protein binding rate: The plasma protein binding rate ranged 73.3 to 76.3% in human, and no sex difference was observed.
Animal study: The distribution of high radio activities was observed in the esophagus, liver, and digestive tract and its contents 15 minutes after single-dose oral administration to rat in the whole-body autoradiogram and tissue distribution studies. Radioactivity was observed in the liver, kidneys, thyroid gland, and the contents of the intestines 168 hours after administration.
Metabolism: In vitro microsomal study has shown that the major isoform of metabolizing enzyme was CYP3A4.
Excretion: Pharmacokinetics was examined after a single intravenous administration of the tritiated nalfurafine hydrochloride in 6 healthy adult male volunteers. The excretion rate of feces and urine was 56.0 and 36.2%, respectively, and the accumulated excretion rate was 92.2% within 14 days after administration. Mainly, the unchanged drug was excreted in the urine while the decyclopropylmethyl form of the product was primarily excreted in the feces. The main metabolite was the decyclopropylmethyl form of the product. The glucuronides were also observed.
A removal efficiency of the product by dialysis was evaluated using four kinds of dialysis membrane. The clearance of the unchanged drug was calculated to be 44.6-61.8 mL/min by conversion with a dialysis membrane area of 1.5 m2. This is a low level compared to the renal clearance of 170-210 mL/min of the unchanged drug in healthy adult male volunteers. However, the unchanged drug was considered to be removed by dialysis regardless of the types of membrane. In addition, metabolites (decyclopropylmethyl form and glucuronides of the product) were also considered to be removed regardless of the types of membrane.
Toxicology: Preclinical safety data: Single-dose toxicity study: Abnormal respiration (tachypnea and bradypnea), unconsciousness and other symptoms were observed in dead mice and rats in single-dose toxicity studies, but no noteworthy changes were observed at necropsy. In the survival animals, the changes in clinical signs had mainly been observed until 2 days after administration, and then disappeared. In rats, the transient body weight loss occurred and then the weight was increased. At necropsy, no changes associated with drug administration were observed.
Repeated-dose toxicity study: The maximum non-lethal oral dose was 50 mg/kg in rats and was 0.3 mg/kg in dogs, confirming that both were well over the anticipated maximum clinical dose (5 μg/body).
No deaths were observed in any of the repeated-dose toxicity studies. Central changes in clinical signs (such as a decrease in locomotor activity), decreased food consumption, decreased body weight gain, and decrease in prostate and seminal vesicle weights were commonly observed in both rats and dogs. All of these symptoms were reversible, and occurred at lower doses in dogs. The NOAEL was also lower for dogs than for rats.
Genotoxicity study: A bacterial reverse mutation study, a chromosome aberration study in cultured mammalian cells, and a micronucleus study in mice were conducted, and all of the studies were negative, showing that nalfurafine hydrochloride is not genotoxic.
A carcinogenicity study: Long-term carcinogenicity study in mice and rats (24-month repeated oral administration) revealed no any carcinogenicity associated with nalfurafine hydrochloride.
Reproduction toxicity studies: There are studies on embryo-fetal development in rats and rabbits. In rats, delayed estrous cycles, decreased fertility rate, decreased mean number of implantations, decreased implantation rate, decreased numbers of live fetuses were observed. NOAELs for the reproductive ability of parent animals and for embryos/fetuses were both concluded to be 1 mg/kg/day.
In rabbits, a trend toward decreases in fetal body weight and placental weight were observed in rabbits. NOAELs for the reproductive ability of parent animals and for embryos/fetuses were thus concluded to be 0.01 mg/kg/day.
No teratogenicity was observed in either rats or rabbits. In a study on pre- and postnatal development, including maternal function, the NOAELs of nalfurafine hydrochloride for general toxicity, reproduction, and fetus development were all considered to be 0.1 mg/kg/day. Specifically, in the 1 mg/kg/day group, decreased fertility rates caused by abnormal parturition, abortion or total sorption of litters were observed in F0 dams, and decreased nursing behavior was also observed in postpartum dams.
In F1 fetuses, low body weight and a tendency toward delayed physical development were observed during the lactation period in the 1 mg/kg/day group. Furthermore, the postnatal survival rate on the 4th day after birth was low, reflecting decreased nursing behavior. Based on these results, administration in pregnant women may adversely affect maternal and child health. (See Use in Pregnancy & Lactation.)
Antigenicity study: In antigenicity studies, the results of several antigenicity studies were all negative. Nalfurafine hydrochloride was considered to have no antigenicity.
Dependence: Although the structure of nalfurafine hydrochloride includes a morphinan skeleton, the results in rats and monkeys suggested that the physical dependence associated with nalfurafine hydrochloride was very weak compare with that associated with morphine, and that nalfurafine hydrochloride is not associated with psychological dependence. A long-term study in patients with chronic liver disease and hemodialysis patients revealed no dependence.
Phototoxicity study: In vitro and in vivo phototoxicity studies were also conducted because the maximum absorption of nalfurafine hydrochloride is at around 280 nm. The results suggested that phototoxicity is unlikely to develop at the anticipated clinical dose (2.5 to 5 μg/body).