For intravenous infusion, following reconstitution. Upon reconstitution a colourless or slightly yellow solution is produced.
Gemcitabine should only be prescribed by a physician qualified in the use of anti-cancer chemotherapy.
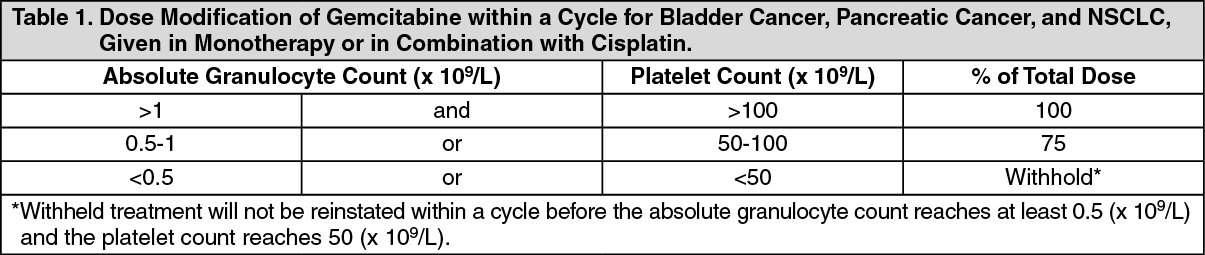
Bladder Cancer (Combination Therapy): Adults: The recommended dose for gemcitabine is 1000 mg/m
2, given as a 30 minute infusion. The dose should be given on days 1, 8, and 15 of each 28 day cycle in combination with cisplatin. Cisplatin is given at a recommended dose of 70 mg/m
2 on day 1 following gemcitabine, or day 2 of each 28 day cycle. This four week cycle is then repeated. Dosage reduction with each cycle or within a cycle may be applied, based upon the amount of toxicity experienced by the patient.
Pancreatic Cancer: Adults: The recommended dose of gemcitabine is 1000 mg/m
2, given by 30 minute intravenous infusion. This should be repeated once weekly for up to 7 weeks, followed by a one week of rest period. Subsequent cycles should consist of gemcitabine infusions once weekly for 3 consecutive weeks out of every four weeks. Dosage reduction with each cycle or within a cycle may be applied, based upon the amount of toxicity experienced by the patient.
Non-Small Cell Lung Cancer (Monotherapy): Adults: The recommended dose of gemcitabine is 1000 mg/m
2, given by 30 minute intravenous infusion. This should be repeated once weekly for three weeks, followed by a one week rest period. This four-week cycle is then repeated. Dosage reduction with each cycle or within a cycle may be applied, based upon the amount of toxicity experienced by the patient.
Non-Small Cell Lung Cancer (Combination Therapy): Adults: The recommended dose of gemcitabine is 1250 mg/m
2, given by 30 minute intravenous infusion, on days 1 and 8 of each 21 day cycle. Dosage reduction with each cycle or within a cycle may be applied, based upon the amount of toxicity experienced by the patient.
Cisplatin has been used at doses between 75-100 mg/m
2 once every 3 weeks.
Ovarian cancer (combination therapy): The recommended dose of gemcitabine, when used in combination with carboplatin, is 1000 mg/m
2, given by 30 minute intravenous infusion, on days 1 and 8 of each 21 day cycle. After gemcitabine, carboplatin will be given on day 1, consistent with a target Area Under the Curve (AUC) of 4.0 mg/mL/min. Dosage reduction with each cycle or within a cycle may be applied, based upon the amount of toxicity experienced by the patient.
Breast cancer (combination therapy): Adults: It is recommended that gemcitabine is used together with paclitaxel according to the following procedure: Paclitaxel (175 mg/m
2) is intravenously infused over 3 hours on day 1, followed by gemcitabine (1250 mg/m
2) intravenously infused for 30 minutes on days 1 and 8 of each 21 day treatment cycle. Dosage reduction with each cycle or within a cycle may be applied based upon the amount of toxicity experienced by the patient. The absolute granulocyte count should be at least 1.5 x 10
9/L before treatment with the gemcitabine + paclitaxel combination.
Monitoring for toxicity and dose modification due to toxicity: Dose adjustment due to non-haematological toxicity: Periodic physical examination and checks of renal and hepatic function should be made to detect non-haematological toxicity. Dosage reduction with each cycle or within cycle may be applied, based upon the amount of toxicity experienced by the patient. In general, for severe (Grade 3 or 4) non-haematological toxicity, except nausea/vomiting, therapy with gemcitabine should be withheld or decreased depending on the judgement of the treating physician. Doses should be withheld until toxicity has been resolved.
For cisplatin, carboplatin, and paclitaxel dosage adjustment in combination therapy, please refer to the corresponding Summary of Product Characteristics.
Dosage Adjustment in the Presence of Haematological Toxicity: Initiation of a cycle: For all indications, patient must be monitored before each dose for platelet and granulocyte counts. Patients should have an absolute granulocyte count of at least 1,500 (x10
6/L) and a platelet count of 100,000 (x10
6/L) prior to administration of a cycle.
Within a cycle: Dose modifications of gemcitabine within a cycle should be performed according to the following tables: (See Tables 1, 2 and 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dose Adjustment Due to Haematological Toxicity in Subsequent Cycles, for All indications:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dose Adjustment Due to Haematological Toxicity in Subsequent Cycles, for All indications: The gemcitabine dose should be reduced to 75% of the original cycle initiation dose, in the case of the following haematological toxicities: Absolute granulocyte count <0.5 x 10
9/L for more than 5 days; Absolute granulocyte count <0.1 x 10
9/L for more than 3 days; Febrile neutropaenia; Platelets <25 x 10
9/L, Cycle delay of more than one week due to toxicity.
Methods of Administration: Gemcitabine is tolerated well during infusion and may be administered ambulant. If extravasation occurs, generally the infusion must be stopped immediately and started again in another blood vessel. The patient should be monitored carefully after the administration.
Special populations: Patients with Hepatic or Renal Impairment: Gemcitabine should be used with caution in patients with hepatic or renal impairment as there is insufficient information from clinical studies to allow for clear dose recommendations for these patient populations (see Precautions and Pharmacology: Pharmacokinetics under Actions).
Elderly Population (>65 years): Gemcitabine has been well tolerated in patients over the age of 65. There is no evidence to suggest that dose adjustments, other than those already recommended for all patients, are necessary in the elderly (see Pharmacology: Pharmacokinetics under Actions).
Paediatric Population (<18 years): Gemcitabine is not recommended for use in children under 18 years of age due to insufficient data on safety and efficacy.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image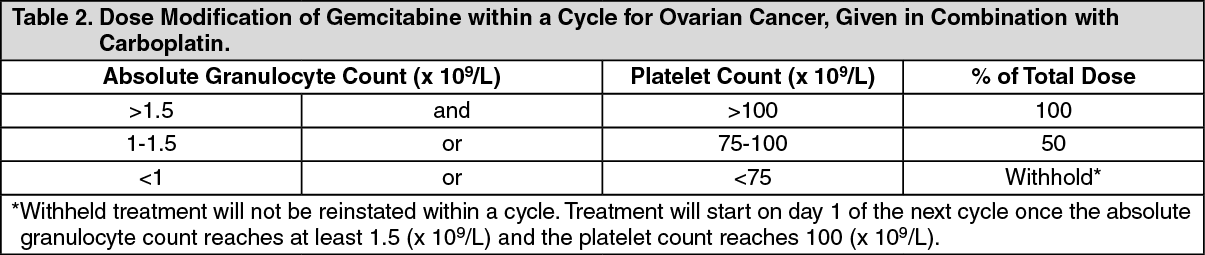 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image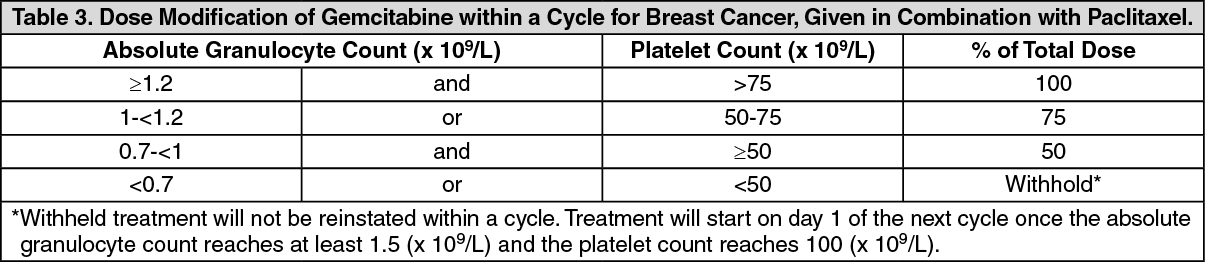 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image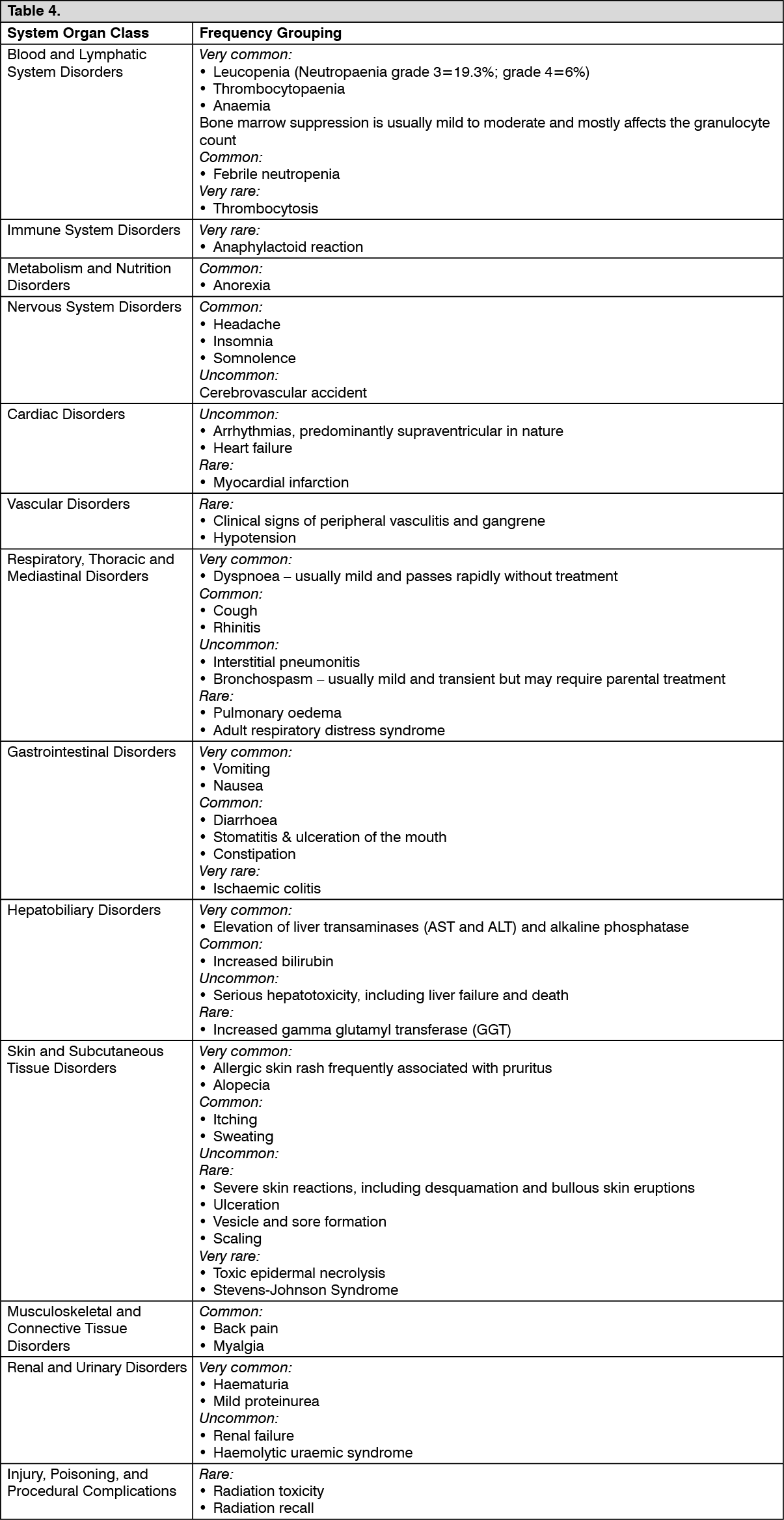 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image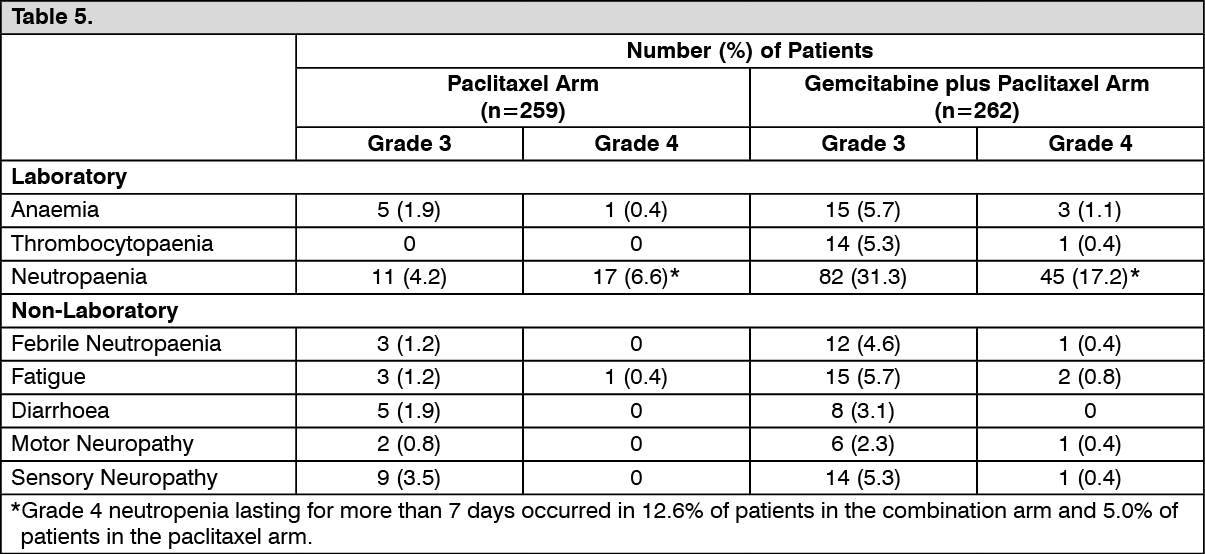 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image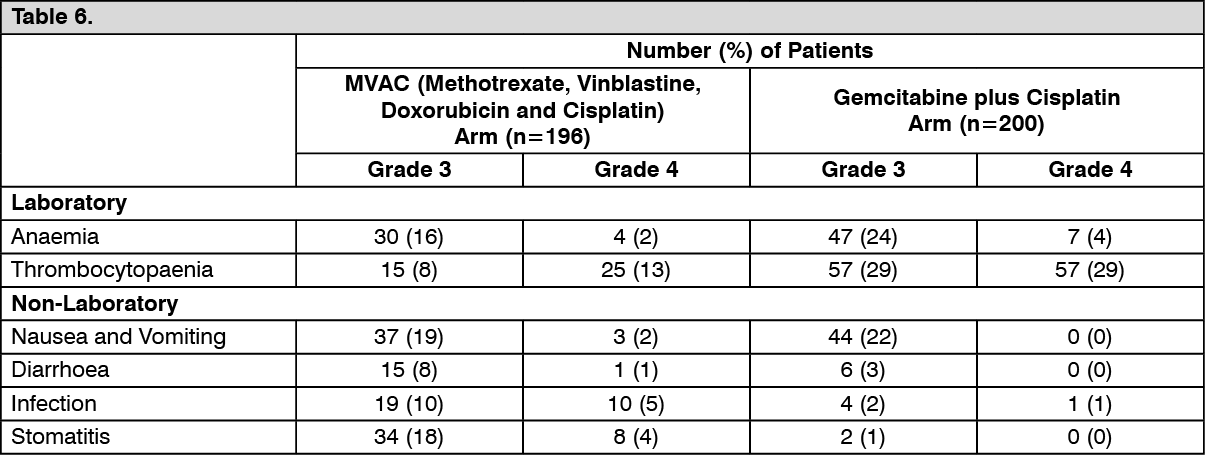 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image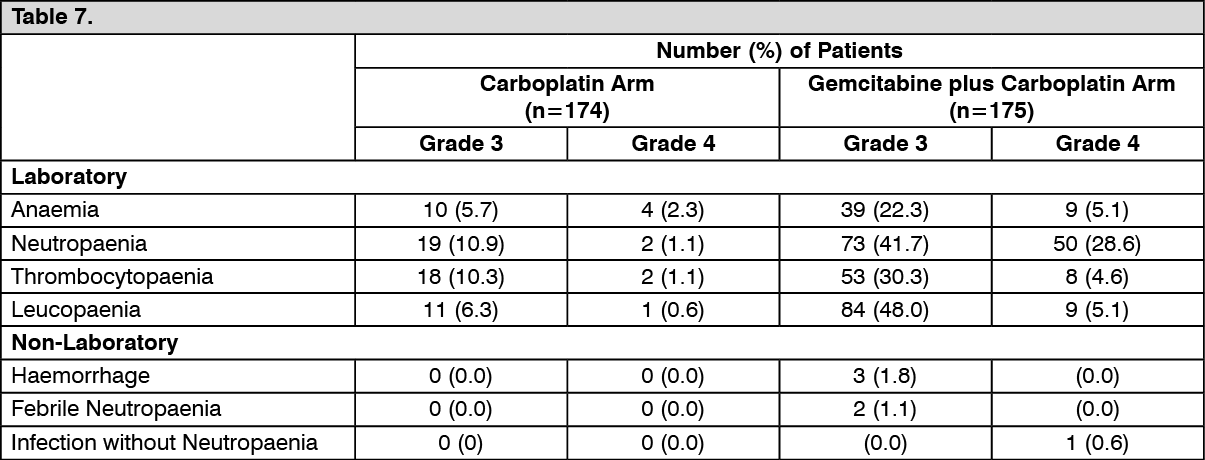 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image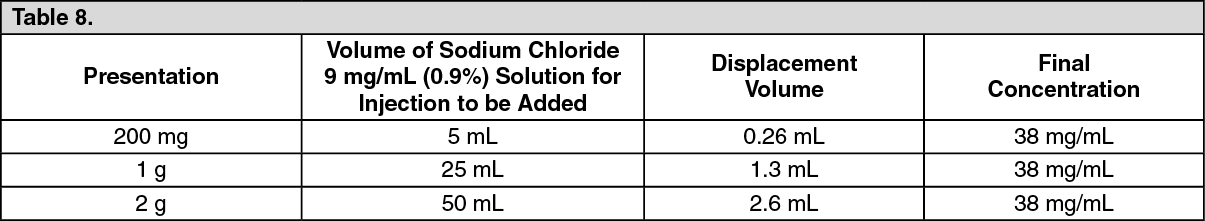 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out