


 Đăng xuất
Đăng xuất
Đơn trị liệu Lynparza có liên quan đến các phản ứng ngoại ý thường ở mức độ nhẹ hoặc trung bình (CTCAE cấp độ 1 hoặc 2) và thường không cần phải ngừng điều trị. Các phản ứng ngoại ý được ghi nhận thường xuyên nhất trong các thử nghiệm lâm sàng ở bệnh nhân dùng đơn trị liệu Lynparza (≥ 10%) là buồn nôn, mệt mỏi, thiếu máu, nôn, tiêu chảy, giảm cảm giác ngon miệng, nhức đầu, rối loạn vị giác, ho, giảm bạch cầu trung tính, khó thở, chóng mặt, khó tiêu, giảm bạch cầu và giảm tiểu cầu.
Các phản ứng ngoại ý cấp độ ≥ 3 xảy ra ở tần suất > 2% bệnh nhân bao gồm thiếu máu (16%), giảm bạch cầu trung tính (6%), mệt mỏi/ suy nhược (6%), giảm bạch cầu (3%), và giảm tiểu cầu (3%).
Các phản ứng ngoại ý thường dẫn đến ngưng tạm thời và/ hoặc giảm liều khi dùng thuốc đơn trị là thiếu máu (16,7%), nôn (6,3%), buồn nôn (6,2%), mệt mỏi/ suy nhược (6,1%) và giảm bạch cầu trung tính (6,0%). Các phản ứng ngoại ý thường gặp nhất dẫn đến ngưng thuốc vĩnh viễn là thiếu máu (1,7%), giảm tiểu cầu (0,8%), mệt mỏi/ suy nhược (0,7%) và buồn nôn (0,7%).
Dữ liệu an toàn thuốc khi dùng phối hợp Lynparza và bevacizumab thường nhất quán với các liệu pháp đơn trị liệu.
Các phản ứng ngoại ý dẫn đến ngưng thuốc tạm thời và/ hoặc giảm liều olaparib khi dùng phối hợp với bevacizumab xảy ra ở 57,4% bệnh nhân và dẫn đến ngưng điều trị vĩnh viễn với olaparib/bevacizumab và giả dược/bevacizumab xảy ra lần lượt ở 20,4% và 5,6% bệnh nhân. Các phản ứng ngoại ý thường gặp nhất dẫn đến ngưng thuốc tạm thời và/ hoặc giảm liều là thiếu máu (20,6%), và buồn nôn (7,5%). Các phản ứng ngoại ý thường gặp nhất dẫn đến ngưng thuốc vĩnh viễn là thiếu máu (3,6%), buồn nôn (3,4%) và mệt mỏi/ suy nhược (1,5%).
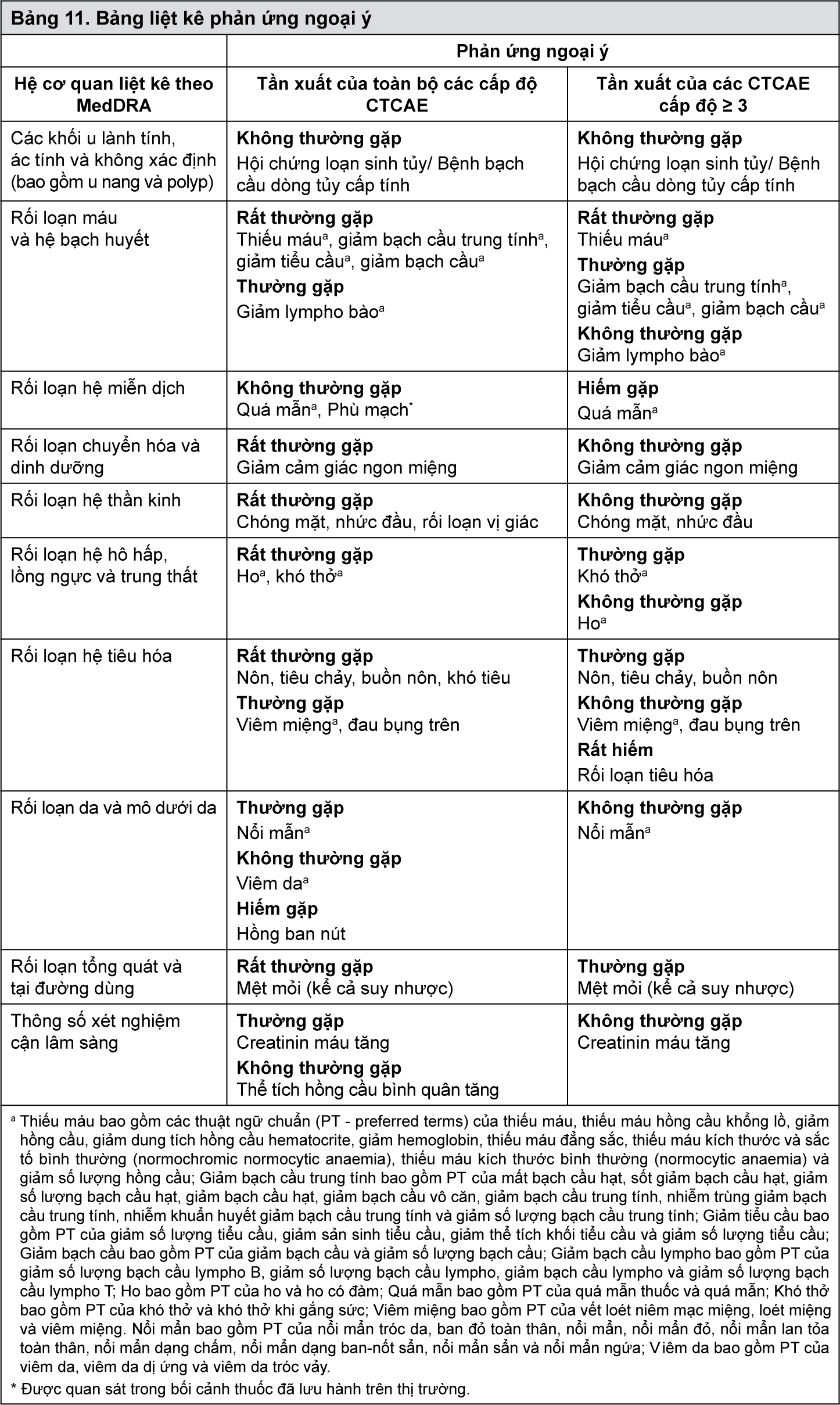
Bảng liệt kê các phản ứng ngoại ý
Hồ sơ an toàn thuốc dựa trên dữ liệu gộp từ 2.901 bệnh nhân với khối u đặc được điều trị bằng đơn trị liệu Lynparza trong các thử nghiệm lâm sàng với liều khuyến cáo.
Các phản ứng ngoại ý sau đây đã được xác định trong các thử nghiệm lâm sàng với bệnh nhân dùng đơn trị liệu Lynparza khi biết mức tiếp xúc trên bệnh nhân. Bảng 11 liệt kê phản ứng ngoại ý của thuốc theo nhóm hệ cơ quan (SOC) trong hệ thống MedDRA và theo thuật ngữ của MedDRA. Trong mỗi hệ cơ quan, các phản ứng ngoại ý được sắp xếp theo tần suất giảm dần và sau đó là mức độ nghiêm trọng giảm. Tần suất xuất hiện các phản ứng ngoại ý được định nghĩa là: rất thường gặp (≥ 1/10); thường gặp (≥ 1/100 đến < 1/10); không thường gặp (≥ 1/1.000 đến < 1/100); hiếm gặp (≥ 1/10.000 đến < 1/1.000); rất hiếm gặp (< 1/10.000); không biết (không thể ước tính từ dữ liệu có sẵn).
- xem Bảng 11.
Mô tả một số phản ứng ngoại ý chọn lọc
Độc tính trên máu
Thiếu máu và các độc tính huyết học khác nói chung ở mức độ thấp (CTCAE cấp độ 1 hoặc 2), tuy nhiên, đã có báo cáo về các biến cố CTCAE cấp độ 3 và cao hơn. Thiếu máu là phản ứng ngoại ý CTCAE cấp độ ≥ 3 phổ biến nhất được ghi nhận trong các nghiên cứu lâm sàng. Thời gian trung bình từ khi dùng thuốc cho đến khi khởi phát thiếu máu đầu tiên là khoảng 4 tuần (khoảng 7 tuần đối với các biến cố CTCAE cấp độ ≥ 3). Thiếu máu được xử trí bằng cách ngưng tạm thời và giảm liều (xem phần Liều lượng và cách dùng) và truyền máu nếu cần. Trong các nghiên cứu lâm sàng trên thuốc dạng viên nén, tần xuất của phản ứng ngoại ý thiếu máu là 39,2% (CTCAE cấp độ ≥ 3 là 17,2%) và tần xuất bệnh nhân ngưng thuốc tạm thời, giảm liều và ngưng thuốc vĩnh viễn do thiếu máu lần lượt là 17,8%, 11,1% và 2,2%; có 21,5% bệnh nhân được điều trị bằng olaparib cần một hoặc nhiều lần truyền máu. Một mối tương quan về mức tiếp xúc - đáp ứng (exposure response relationship) giữa olaparib và việc giảm hemoglobin đã được chứng minh. Trong các nghiên cứu lâm sàng với Lynparza, tần xuất CTCAE cấp độ ≥ 2 (giảm) so với ban đầu ở hemoglobin là 20%, số lượng bạch cầu trung tính 20%, tiểu cầu 5%, bạch cầu lympho 30% và bạch cầu 20% (tất cả tỷ lệ % đều là xấp xỉ).
Tần suất tăng thể tích hồng cầu (MCV) trung bình từ thấp hoặc bình thường lúc đầu đến > ULN (ngưỡng giới hạn cao - upper limit of normal) là khoảng 68%. Các mức độ dường như sẽ trở lại bình thường sau khi ngừng điều trị và không có bất kỳ hậu quả lâm sàng nào.
Kiểm tra công thức máu tổng quát trước khi điều trị và theo dõi hàng tháng sau đó được khuyến cáo trong 12 tháng đầu điều trị và định kỳ sau thời gian này để theo dõi những thay đổi đáng kể về lâm sàng của bất kỳ tham số nào trong quá trình điều trị mà điều này có thể cần phải ngừng tạm thời hoặc giảm liều và/ hoặc điều trị thêm (xem phần Liều lượng và cách dùng và Cảnh báo và thận trọng).
Hội chứng loạn sinh tủy/Bệnh bạch cầu dòng tủy cấp tính
MDS/AML là phản ứng ngoại ý nghiêm trọng xuất hiện với tần suất không thường gặp (0,4%) trong các nghiên cứu lâm sàng khi dùng liệu pháp đơn trị liệu ở tất cả các chỉ định của thuốc. Tần xuất 0,5% bao gồm các biến cố được báo cáo trong thời gian theo dõi an toàn lâu dài (tỷ lệ được tính toán dựa trên tổng dân số an toàn từ 16.108 bệnh nhân điều trị ít nhất một liều olaparib đường uống trong các nghiên cứu lâm sàng). Tất cả các bệnh nhân có các yếu tố nguy cơ tiềm năng phát triển MDS/AML, đều đã được hóa trị liệu trước đó với các tác nhân chứa platinum. Nhiều bệnh nhân cũng đã được điều trị bằng các tác nhân phá hủy DNA khác và xạ trị. Phần lớn các báo cáo là ở bệnh nhân ung thư vú có đột biến gen BRCA 1 hoặc 2 dạng di truyền (germline) (gBRCA1/2). Tỷ lệ mới mắc MDS/AML là tương tự nhau giữa bệnh nhân có đột biến gen BRCA1 dạng di truyền (germline) và bệnh nhân có đột biến gen BRCA2 dạng di truyền (germline) (lần lượt là 2,3% và 1,6%). Một số bệnh nhân có tiền sử ung thư trước đây hoặc mắc chứng loạn sản tủy xương.
Đối với bệnh nhân ung thư buồng trứng tái phát nhạy với platinum có đột biến BRCA mà đã được điều trị ít nhất 2 bước hóa trị liệu chứa platinum và được điều trị olaparib cho đến khi bệnh tiến triển (nghiên cứu SOLO2, 45% bệnh nhân dùng olaparib ≥ 2 năm), với thời gian theo dõi 5 năm tần xuất xuất hiện MDS/AML là 8,2% ở bệnh nhân điều trị bằng olaparib và 4% ở nhóm bệnh nhân dùng giả dược. Ở nhánh olaparib, 9 trong 16 trường hợp MDS/AML xuất hiện sau khi ngừng điều trị olaparib, trong giai đoạn theo dõi sống còn toàn bộ. Tần xuất MDS/AML được ghi nhận trong trong giai đoạn theo dõi kéo dài về sống còn toàn bộ ở nhánh olaparib và khởi phát muộn MDS/AML. Nguy cơ MDS/AML duy trì mức < 1,5% tại thời điểm 5 năm theo dõi trong điều trị duy trì bước 1 bằng olaparib trong thời gian 2 năm sau đợt hóa trị liệu có chứa platinum (là 1,2% trong nghiên cứu SOLO1 và 0,7% trong nghiên cứu PAOLA-1). Để kiểm soát và hạn chế nguy cơ, xem phần Cảnh báo và thận trọng.
Các xét nghiệm cận lâm sàng khác
Trong các nghiên cứu lâm sàng với Lynparza, tần xuất gặp biến cố ngoại ý CTCAE cấp độ ≥ 2 (tăng) từ trị số creatinine máu ban đầu là khoảng 11%. Dữ liệu từ một nghiên cứu mù đôi có đối chứng giả dược cho thấy giá trị trung bình tăng 23% so với ban đầu duy trì theo thời gian và trở lại mức ban đầu sau khi ngừng điều trị, không có hậu quả rõ rệt trên lâm sàng. 90% bệnh nhân có giá trị creatinine CTCAE cấp độ 0 ở thời điểm ban đầu và 10% bệnh nhân có CTCAE cấp độ 1 ở thời điểm ban đầu.
Độc tính hệ tiêu hóa
Buồn nôn thường được ghi nhận rất sớm, phần lớn bệnh nhân sẽ khởi phát trong tháng đầu điều trị Lynparza. Nôn mửa đã được báo cáo sớm với cơn khởi phát đầu tiên xuất hiện ở phần lớn bệnh nhân trong vòng hai tháng đầu điều trị Lynparza. Cả hai triệu chứng buồn nôn và nôn được ghi nhận không liên tục trên phần lớn bệnh nhân và có thể được kiểm soát bằng cách ngưng tạm thời thuốc, giảm liều và/ hoặc điều trị chống nôn. Không cần thiết phải dự phòng chống nôn.
Trong điều trị duy trì bước 1 ung thư buồng trứng, tần xuất bệnh nhân gặp triệu chứng buồn nôn (77% khi dùng olaparib, 38% khi dùng giả dược), nôn (40% khi dùng olaparib, 15% khi dùng giả dược), tiêu chảy (34% khi dùng olaparib, 25% khi dùng giả dược) và khó tiêu (17% trên olaparib, 12% trên giả dược). Các biến cố buồn nôn dẫn đến ngừng thuốc xảy ra ở 2,3% bệnh nhân điều trị bằng olaparib (CTCAE cấp độ 2) và ở 0,8% bệnh nhân được điều trị bằng giả dược (CTCAE độ 1); có 0,8% và 0,4% bệnh nhân điều trị bằng olaparib phải ngừng điều trị do nôn mửa và khó tiêu ở cấp độ thấp (CTCAE độ 2). Không có bệnh nhân điều trị bằng olaparib hoặc giả dược phải ngừng điều trị do tiêu chảy. Không có bệnh nhân điều trị bằng giả dược phải ngưng dùng do nôn mửa hoặc khó tiêu. Các biến cố buồn nôn dẫn đến ngưng thuốc tạm thời và giảm liều ở 14% và 4% tương ứng ở những bệnh nhân được điều trị bằng olaparib. Các biến cố nôn mửa dẫn đến ngưng thuốc tạm thời xảy ra trên 10% bệnh nhân dùng olaparib; không có bệnh nhân điều trị bằng olaparib gặp biến cố nôn mửa dẫn đến giảm liều.
Trẻ em
Không có nghiên cứu tiến hành trên trẻ em.
Các nhóm bệnh nhân khác
Rất ít dữ liệu về tính an toàn trên bệnh nhân không phải là người da trắng.
Báo cáo phản ứng ngoại ý nghi ngờ do thuốc
Báo cáo các phản ứng ngoại ý nghi ngờ do thuốc sau khi thuốc được cấp phép lưu hành là quan trọng. Điều này cho phép tiếp tục theo dõi sự cân bằng giữa lợi ích/nguy cơ của thuốc. Cán bộ y tế được yêu cầu báo cáo bất kỳ tác dụng không mong muốn nào qua hệ thống báo cáo quốc gia.
View ADR Monitoring Form